
The landscape of modern medicine has been profoundly shaped by covalent drugs, a class of therapeutics whose impact dates back over a century to the approval of aspirin in 1899. This initial breakthrough was swiftly followed by penicillin, a discovery that fundamentally altered the course of infectious disease treatment and remains one of the most significant pharmaceutical advancements in human history. In recent decades, particularly over the last ten years, the field has experienced a remarkable resurgence, marked by an increasing number of novel covalent drugs gaining regulatory approval across a diverse spectrum of conditions. This renaissance offers renewed hope for patients battling diseases previously considered "undruggable," opening new avenues for therapeutic intervention. However, the development of these potent agents is not without its complexities, primarily stemming from their unique pharmacokinetic (PK) characteristics, which necessitate specialized strategies throughout the drug discovery and development pipeline.
A Historical Perspective: From Serendipity to Strategic Design
The early triumphs of covalent drugs were largely serendipitous, with their precise mechanisms of action remaining a mystery for decades after their market introduction. Aspirin, for instance, was approved for its analgesic and anti-inflammatory properties long before its mechanism of inhibiting cyclooxygenase (COX) activity through covalent bond formation was fully elucidated in 1971, nearly 70 years post-approval. Similarly, the covalent inhibition mechanism of penicillin, which disrupts bacterial cell wall synthesis by binding to bacterial transpeptidase enzymes, was not understood until over 50 years after its widespread clinical use. These early discoveries laid the groundwork, revealing that covalent drugs exert their effects by forming stable, irreversible covalent bonds with biological macromolecules, such as enzymes or receptors, thereby permanently altering or deactivating their function. Penicillin’s efficacy, for example, arises from its ability to covalently acylate a serine residue within the active site of bacterial transpeptidases, crucial enzymes involved in peptidoglycan cross-linking, ultimately leading to bacterial cell death.
Despite these early successes, the development of new covalent drugs largely stalled for a significant period. The primary deterrent was a pervasive concern regarding the safety profile of the electrophilic compounds typically employed in their design. Early toxicological studies revealed that certain chemicals could be metabolized in vivo into highly reactive electrophiles. These reactive species possessed the unfortunate propensity to bind indiscriminately to a wide array of endogenous biomolecules, including proteins, lipids, and even nucleic acids like DNA, leading to widespread cellular damage, off-target toxicities, and, in some cases, genotoxicity and carcinogenesis. High-profile instances of such toxicities, exemplified by the hepatotoxicity associated with acetaminophen overdose, the liver damage induced by bromobenzene, and the severe dermatological reactions caused by urushiol (the active irritant in poison ivy), solidified the perception that electrophiles represented "no-go" zones for drug development. Consequently, medicinal chemistry efforts largely shifted towards the pursuit of reversible, non-covalent inhibitors, a strategy perceived as inherently safer, and the covalent approach was largely avoided for several decades.
The Dawn of Precision: Modern Covalent Drug Design
The modern era of covalent drug development has been ushered in by a fundamental re-evaluation of electrophiles and a sophisticated understanding of their reactivity. Contemporary covalent drugs now leverage "soft electrophiles," characterized by their carefully tuned reactivity and selectivity. These include chemical moieties such as acrylamides, nitriles, chloroacetamides, and sulfonyl fluorides, often referred to as "warheads." Unlike their highly reactive predecessors, these newer electrophiles are designed to be far less promiscuous and have demonstrated remarkable safety and clinical success, effectively dispelling the earlier belief that all electrophilic compounds are inherently promiscuous and unsafe.
This critical shift underscores the paramount importance of meticulously tuning the electrophilicity of the warhead during the early stages of drug discovery. The goal is to achieve sufficient reactivity for target engagement while simultaneously minimizing reactivity towards off-target biomolecules, thereby reducing systemic toxicity. A stark illustration of this principle can be found in the comparison between sucralose, an artificial sweetener, and sulfur mustard, a potent chemical weapon. Both compounds contain alkyl chloride functionalities. However, sucralose is safely consumed long-term as a food additive, whereas sulfur mustard causes severe alkylation damage due to its exceptionally high reactivity. The precise control over reactivity is what distinguishes a safe therapeutic from a toxic agent.
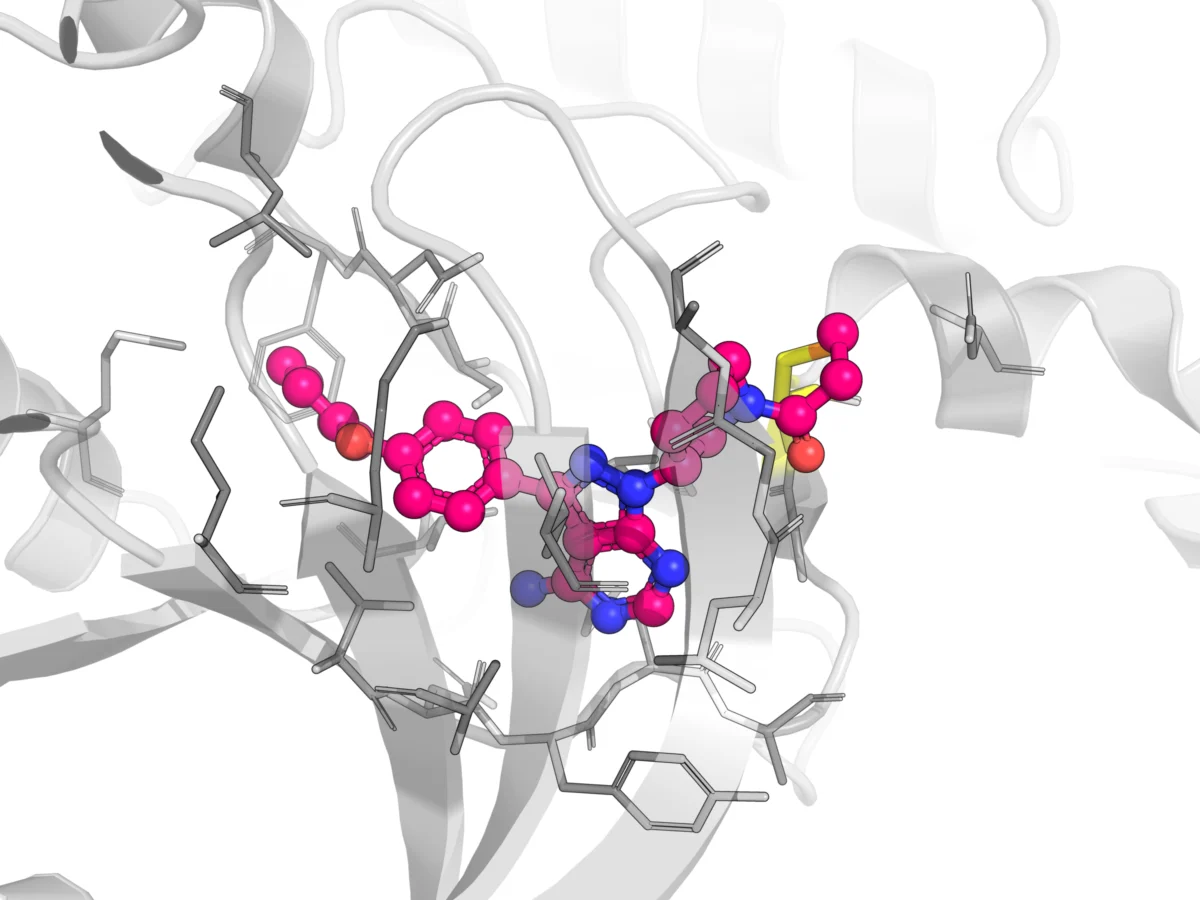
The success of these lower-reactivity warheads, particularly when integrated into rationally designed Targeted Covalent Inhibitors (TCIs), has ignited a robust boom in covalent drug development. TCIs are engineered to selectively target specific nucleophilic residues (most commonly cysteine, but also lysine or serine) within the active site or an allosteric pocket of a disease-relevant protein. This targeted approach ensures that the covalent bond formation is highly specific, leading to potent and durable inhibition. To date, more than 110 covalent drugs have received regulatory approval across a multitude of therapeutic areas, showcasing their broad applicability. Beyond penicillin, notable examples include omeprazole for gastrointestinal disorders, telaprevir for hepatitis C virus (HCV) infection, and osimertinib for oncology (specifically non-small cell lung cancer with EGFR T790M mutation). A prime example of a highly successful TCI is ibrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase (BTK), which has transformed the treatment landscape for various B-cell malignancies and certain autoimmune diseases. Ibrutinib’s acrylamide warhead is precisely positioned to form a covalent bond with Cys481 in the ATP-binding pocket of BTK, a structural basis for its prolonged target occupancy and profound pharmacological effect.
As the field has matured, researchers have adopted a more sophisticated array of metrics for evaluating covalent drugs. These now extend beyond traditional binding affinities to include kinetic parameters that account for their reaction rates and potential off-target effects. Key metrics include kinact/Ki, which quantifies the efficiency of irreversible enzyme inactivation, assessment of glutathione (GSH) conjugation as an indicator of electrophilic reactivity and potential for metabolic clearance, and time-dependent inhibition (TDI) risk assessment for cytochrome P450 (CYP450) enzymes. While these advancements have significantly improved the predictability and safety of covalent drugs, their unique mechanisms inherently present lingering pharmacokinetic challenges that demand meticulous consideration.
Navigating the Unique Pharmacokinetic Landscape
The fundamental principle of covalent binding mechanism gives rise to pharmacokinetic properties that distinctly diverge from those of conventional, reversible small-molecule drugs. Key considerations that demand specialized attention include protein binding dynamics, distinct clearance mechanisms, the potential for time-dependent inhibition of metabolizing enzymes, and a fundamentally altered understanding of the pharmacokinetics/pharmacodynamics (PK/PD) relationship.
A. Beyond the Target: Protein Binding and Distribution
While covalent binding to the intended therapeutic target protein is the cornerstone of a covalent drug’s efficacy, these agents can also form covalent adducts with other proteins throughout the body. These off-target interactions are not merely benign; they can significantly influence the drug’s systemic distribution, metabolic fate, and overall toxicity profile. For instance, covalent drugs frequently form stable adducts with highly abundant plasma proteins, most notably human serum albumin (HSA). Such binding can effectively reduce the concentration of free, unbound drug in the plasma, which in turn influences its distribution to target tissues, its metabolic clearance, and its potential for renal excretion. While high levels of plasma protein binding might initially seem problematic, it does not inherently correlate with adverse effects. In some cases, HSA binding can act as a circulating reservoir, slowly releasing the drug and prolonging its systemic exposure.
Beyond plasma, covalent drugs can also bind to various tissue proteins, particularly in highly metabolically active organs like the liver. This tissue binding can lead to drug retention, potentially impacting local metabolism, and contributing to organ-specific toxicity if the off-target interactions are extensive or involve critical cellular proteins. Therefore, drug developers face the critical task of distinguishing beneficial, on-target engagement from detrimental, off-target reactivity before attributing toxicity. Advice for Developers: A rigorous and iterative process of optimizing the selectivity and reactivity of these inhibitors is paramount. This involves not only designing warheads with appropriate electrophilicity but also ensuring the overall molecular architecture guides the drug preferentially to its intended target, minimizing undesirable interactions with bystander proteins.
B. The Complexities of Clearance: Conjugation and Adduct Turnover
A distinctive feature of many covalent drugs is their reliance on specific conjugation pathways for clearance, particularly the formation of glutathione (GSH) and cysteine adducts. These pathways represent major routes of metabolic inactivation for electrophilic compounds. Glutathione S-transferases (GSTs) are a family of enzymes that catalyze the conjugation of electrophilic compounds with the endogenous nucleophile glutathione, rendering them more water-soluble and amenable to excretion. Non-enzymatic conjugation with GSH or other nucleophilic species (like cysteine) can also occur.
Consider Futibatinib, an irreversible FGFR1-4 inhibitor recently approved for the treatment of FGFR2-rearranged intrahepatic cholangiocarcinoma. Its primary metabolic pathways include O-demethylation and extensive glutathione conjugation. A detailed mass-balance study involving 14C-futibatinib revealed that a major circulating metabolite was a cysteinylglycine conjugate, accounting for approximately 13% of the circulating drug-related material. Further in vitro hepatocyte studies identified additional metabolites, including GSH, cysteine, glucuronide, and sulfate conjugates, highlighting the complexity of its metabolic disposition.
Beyond direct conjugation, covalent drugs can also be eliminated through the breakdown and clearance of drug-protein adducts, such as those formed with albumin. This "protein-adduct mediated clearance" mechanism can account for a significant portion of the total drug excretion, especially for drugs that form stable, long-lived adducts with circulating proteins. The turnover rate of the adducted protein then dictates the rate of drug elimination from the system. Advice for Developers: It is imperative for development teams to quantify both nonenzymatic and GST-mediated reactivity early in the discovery process. This allows for the ranking of electrophile reactivity across different lead compounds. Furthermore, utilizing recombinant GST isoforms and hepatocyte systems is crucial for assessing potential polymorphism-related risks, as genetic variations in GST enzymes can significantly alter an individual’s capacity to metabolize these drugs. Understanding these pathways is vital for predicting human pharmacokinetics and potential drug-drug interactions.
C. Time-Dependent Inhibition (TDI) of Cytochrome P450 Enzymes
Another critical pharmacokinetic consideration for covalent drugs is their propensity to act as time-dependent inhibitors (TDIs) of cytochrome P450 (CYP450) enzymes. CYP450 enzymes are a superfamily of monooxygenases primarily responsible for the metabolism of the vast majority of xenobiotics, including many drugs. TDI occurs when a drug, after being metabolized by a CYP enzyme, forms a reactive intermediate that then irreversibly binds to and inactivates the same or another CYP enzyme. This leads to a progressive and sustained decrease in the enzyme’s metabolic capacity over time, beyond what would be predicted by simple competitive inhibition.
Studies have consistently shown that a significant proportion of covalent drugs exhibit time-dependent inhibition of at least one human CYP enzyme isoform. For example, a seminal study by Moghaddam MF et al. (2014) evaluated ten diverse covalent drugs across multiple CYP isoforms and found that the majority demonstrated time-dependent inhibition of at least one enzyme. This observation strongly suggests that many covalent drugs inherently carry a risk of TDI, which can lead to clinically significant drug-drug interactions (DDIs) when co-administered with other medications that are substrates for the inhibited CYP enzyme. Such DDIs can result in elevated plasma concentrations of co-administered drugs, potentially leading to increased toxicity or altered therapeutic effects. Advice for Developers: Early and thorough kinetic characterization of potential TDI (including determination of KI, the inhibition constant, and kinact, the maximum rate of inactivation) is essential. Mechanistic modeling of TDI can then be used to predict the clinical DDI risk, which can guide structural optimization efforts to mitigate this risk or inform appropriate dosing strategies and patient monitoring plans during clinical development.
D. Decoupling PK and PD: A Paradigm Shift in Efficacy Assessment
Perhaps the most unique and challenging aspect of covalent drugs pertains to their relationship between pharmacokinetics (plasma drug concentration) and pharmacodynamics (pharmacological effect). For conventional, reversible small-molecule drugs, continuous pharmacological effect typically necessitates maintaining blood concentrations within a specific therapeutic window, as the drug’s effect is directly proportional to its unbound concentration at the target site. However, for covalent drugs, once the irreversible bond is formed with the target protein, the drug persistently occupies the active site, exerting its effect regardless of subsequent plasma drug concentrations. The duration of this pharmacological effect is then dictated not by the drug’s plasma half-life, but by the biological half-life of the target protein itself—specifically, the time required for the cell to synthesize new, uninhibited target protein.
This fundamental decoupling of PK and PD is vividly illustrated by several clinical examples. For instance, with the irreversible BTK inhibitor CC-292, plasma drug levels were observed to fall to near or even below the lower limit of quantification within 24 hours post-dose in clinical trials. Yet, target occupancy of BTK remained remarkably high for up to 24 hours and only began to decline as new BTK protein was synthesized by the cells. Similarly, as depicted in the introductory image, ibrutinib’s prolonged target occupancy of BTK persists long after its plasma concentrations have diminished, demonstrating that plasma PK alone is an inadequate predictor of its pharmacological effect.
This critical distinction implies that for covalent drugs, optimizing systemic plasma exposure over prolonged periods is less critical than achieving rapid and sufficient initial target engagement. The goal shifts from maintaining a steady-state plasma concentration to ensuring a rapid and high maximum concentration (Cmax) that facilitates complete and irreversible binding to the target. Once the target is fully occupied, subsequent plasma drug concentrations become largely irrelevant until the target protein needs to be replenished. Advice for Developers: To effectively navigate this unique PK/PD relationship, researchers should pivot their focus towards achieving Cmax-driven rapid engagement. It is imperative to directly model target occupancy, rather than relying solely on plasma drug concentrations, as the pharmacodynamic effect is fundamentally governed by the turnover rate of the target protein. This often necessitates the development and utilization of sophisticated in vivo and ex vivo assays to directly measure target engagement and protein resynthesis rates in relevant biological matrices.
The Broader Impact and Future Outlook
Covalent drugs represent a powerful and increasingly vital modality in addressing some of the most critical unmet medical needs in modern therapeutics. Their ability to achieve durable target inhibition, often at lower doses and with less frequent administration compared to reversible counterparts, holds immense promise for improving patient outcomes and quality of life, particularly in chronic conditions like cancer and autoimmune diseases. To fully capitalize on this transformative potential, drug developers must cultivate a deep and nuanced understanding of their unique pharmacokinetic properties and apply this knowledge effectively throughout every stage of development, from early discovery to regulatory review.
Successful covalent drug programs must unequivocally demonstrate deliberate and selective chemistry, providing a clear mechanistic understanding of both their efficacy and safety profiles. This includes robust data on target engagement kinetics, off-target reactivity, metabolic pathways, and the distinct PK/PD relationship. As technology continues its relentless march forward, advancements in structural biology, computational chemistry, in situ reaction monitoring, and sophisticated in vitro and in vivo pharmacology will further accelerate the pace of covalent drug discovery and development. Development teams equipped with the specialized expertise and extensive experience required to design, characterize, and accurately model these unique mechanisms will be optimally positioned to efficiently and safely bring truly transformative therapies to patients who desperately need them. The covalent drug renaissance is not merely a scientific curiosity; it is a critical frontier in precision medicine, demanding precision in its development.
SOURCES:
- Moghaddam MF, Tang Y, O’Brien Z, Richardson SJ, Bacolod M, Chaturedi P, Apuy J, Kulkarni A. A proposed screening paradigm for discovery of covalent inhibitor drugs. Drug Metab Lett. 2014;8(1):19-30.
- Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, Witowski SR, Lounsbury H, Chaturvedi P, Mazdiyasni H, Zhu Z, Nacht M, Freed MI, Petter RC, Dubrovskiy A, Singh J, Westlin WF. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013 Aug;346(2):219-28.









Leave a Reply