
Covalent drugs represent a transformative advancement in medicine, offering unprecedented therapeutic efficacy by forming stable, irreversible bonds with their biological targets. From the early serendipitous discoveries of aspirin in 1899 and penicillin shortly thereafter, which profoundly reshaped healthcare, to a recent surge in rationally designed compounds, these agents are now addressing a wide spectrum of diseases, including previously "undruggable" conditions. However, their unique mechanism of action inherently leads to distinct pharmacokinetic (PK) properties that diverge significantly from conventional reversible drugs, presenting developers with specialized challenges in areas such as protein binding, clearance mechanisms, drug-drug interactions, and the critical pharmacokinetic/pharmacodynamic (PK/PD) relationship. Navigating these complexities is paramount for optimizing drug design, ensuring patient safety, and achieving successful regulatory approval in this rapidly evolving field.
A Storied Past and a Modern Renaissance
The journey of covalent drugs has been marked by periods of both pioneering success and cautious skepticism. Early breakthroughs, while revolutionary, were largely empirical, with the precise molecular mechanisms of action often remaining elusive for decades. Aspirin, for instance, a cornerstone of pain relief and anti-inflammatory therapy, was approved long before its ability to covalently inhibit cyclooxygenase (COX) enzymes was elucidated in the 1970s. Similarly, penicillin, discovered in 1928 and mass-produced by the 1940s, halted bacterial infections by forming a covalent bond with a serine residue on bacterial transpeptidase enzymes, a mechanism understood only much later. These early agents demonstrated the immense therapeutic potential of irreversible target engagement, yet the underlying principles guiding their design and safety were largely undeveloped.
By the mid-20th century, the pharmaceutical industry began to steer away from covalent drug development. This shift was largely driven by a growing understanding of toxicology and the perceived inherent dangers of electrophilic compounds, which are fundamental to covalent bond formation. Many early electrophiles were found to react indiscriminately with a multitude of biological macromolecules, including proteins, lipids, and even DNA, leading to off-target toxicities, cellular damage, and in some cases, carcinogenicity. High-profile examples of severe adverse drug reactions associated with compounds like acetaminophen (hepatic toxicity from a reactive metabolite), bromobenzene, and the potent vesicant urushiol (from poison ivy) cemented the notion that electrophiles were "no-go" zones in medicinal chemistry. Consequently, research and development efforts predominantly focused on designing reversible, non-covalent small-molecule inhibitors, which offered a seemingly more controllable and safer pharmacological profile. This era of avoidance lasted for several decades, creating a significant gap in the exploration of covalent strategies.
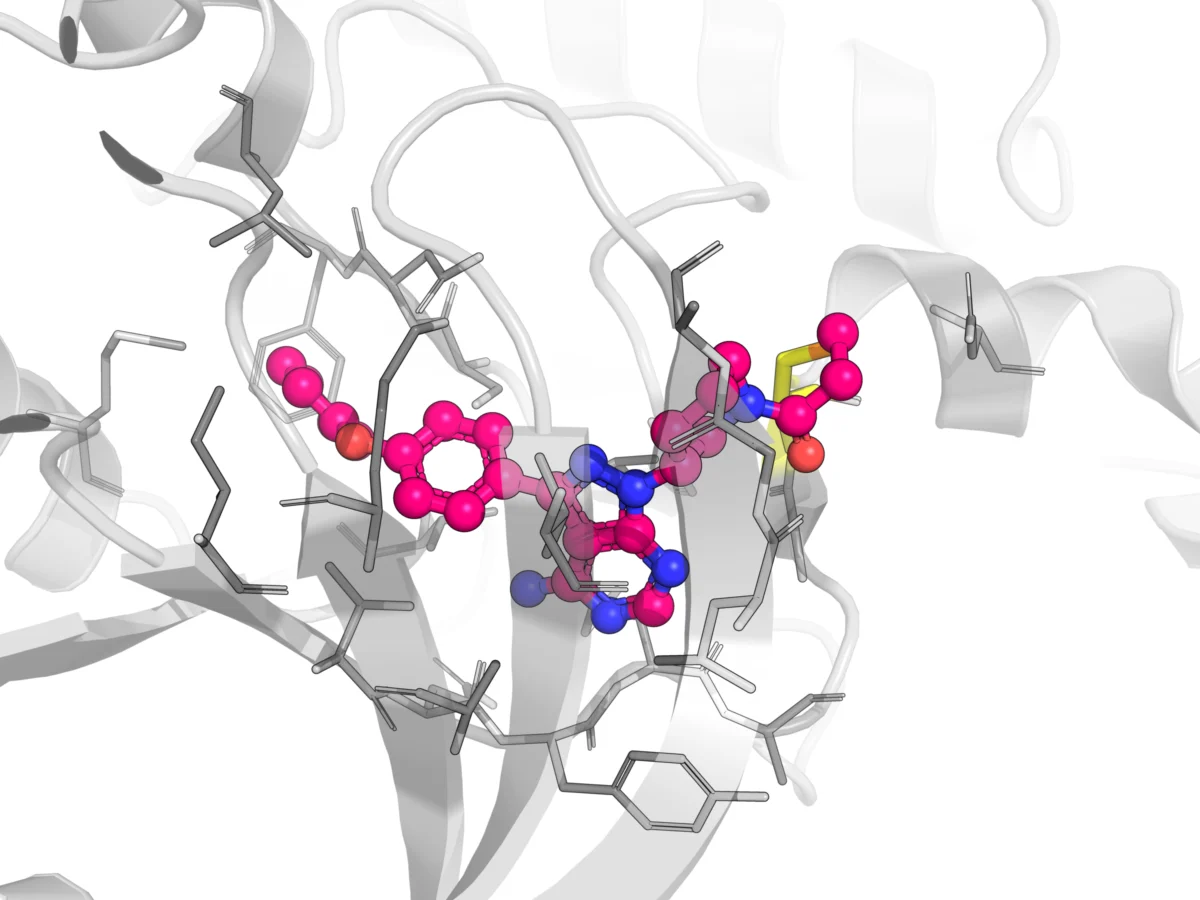
The landscape began to shift dramatically in the early 21st century, ushering in what many refer to as a "renaissance" in covalent drug discovery. This resurgence was fueled by significant advancements in structural biology, computational chemistry, and synthetic methodologies, which enabled the rational design of Targeted Covalent Inhibitors (TCIs). The key innovation was the development of "soft electrophiles"—functional groups like acrylamides, nitriles, and sulfonyl fluorides—that possess carefully tuned reactivity. Unlike their highly promiscuous predecessors, these warheads are designed to react selectively with specific nucleophilic residues (most commonly cysteine thiols, but also lysines, serines, and histidines) within the active site of a target protein. This precise targeting minimizes off-target reactions, significantly enhancing both efficacy and safety.
The success of this rational design approach is evident in the increasing number of covalent drugs approved since 2010. Over 110 covalent drugs are now on the market, spanning diverse therapeutic areas. Notable modern examples include:
- Ibrutinib (Imbruvica): An irreversible inhibitor of Bruton’s tyrosine kinase (BTK), approved for various B-cell malignancies, showcasing the power of TCIs in oncology. Its acrylamide warhead forms a covalent bond with Cys481 in BTK, leading to prolonged target occupancy despite rapid plasma clearance.
- Osimertinib (Tagrisso): A third-generation EGFR inhibitor for non-small cell lung cancer (NSCLC) with specific T790M resistance mutations, utilizing a similar targeted covalent strategy.
- Omeprazole (Prilosec): A proton pump inhibitor for gastrointestinal diseases, which covalently inhibits H+/K+-ATPase.
- Telaprevir (Incivek): An HCV protease inhibitor that, while later withdrawn due to side effects, demonstrated the efficacy of covalent binding in antiviral therapy.
The market for targeted therapies, including TCIs, has seen robust growth, driven by their enhanced selectivity and sustained pharmacological effect. Investment in this area has escalated, with numerous biotech startups and major pharmaceutical companies actively pursuing covalent strategies, particularly in oncology, autoimmune disorders, and infectious diseases. This paradigm shift underscores the importance of a nuanced understanding of the delicate balance between reactivity and selectivity, transforming what was once considered a liability into a powerful therapeutic advantage.
The Pharmacokinetic Conundrum: Navigating Unique Properties
Despite their therapeutic promise, the irreversible nature of covalent binding introduces distinct pharmacokinetic challenges that demand specialized consideration throughout the drug development pipeline. The conventional PK metrics and models designed for reversible drugs often fail to accurately predict the in vivo behavior and pharmacological effect of covalent inhibitors. Key areas requiring particular attention include protein binding beyond the target, complex clearance mechanisms, time-dependent enzyme inhibition, and the fundamental decoupling of plasma concentration from pharmacodynamic effect.
1. Beyond the Target: Off-Target Protein Binding and Distribution
While selective covalent binding to the intended therapeutic target is crucial for efficacy, covalent drugs can also form adducts with other proteins in the body, influencing their distribution, metabolism, and potential for toxicity. These off-target interactions are not always detrimental but require careful characterization.
One significant interaction involves plasma proteins, particularly human serum albumin (HSA). Covalent drugs can form stable adducts with HSA, a phenomenon that can dramatically reduce the concentration of free, unbound drug in plasma. This reduction in free drug can impact distribution to target tissues and influence systemic clearance rates. While high levels of plasma protein binding are common for many drugs, covalent binding to non-target proteins adds a layer of complexity. The formation of these adducts can act as a circulating reservoir, potentially prolonging the drug’s systemic exposure, or it can facilitate unique clearance pathways.
Beyond plasma, covalent drugs can also bind to tissue proteins, particularly in organs like the liver, kidney, and gastrointestinal tract. Such tissue-specific covalent binding can affect the drug’s local concentration, retention time within the tissue, and metabolic fate, potentially contributing to organ-specific toxicities or, conversely, localized therapeutic effects. For instance, high liver binding could either increase the risk of hepatotoxicity or enhance the drug’s action against liver-resident pathogens or cancer cells.
Advice for Developers: A critical step for developers is to meticulously distinguish between on-target engagement, which is essential for therapeutic effect, and off-target reactivity, which can lead to adverse events. Advanced in vitro and in vivo methodologies, including mass spectrometry-based proteomics, covalent adductomic profiling, and sophisticated imaging techniques, are crucial for identifying and quantifying both target and off-target covalent binding. This allows for a comprehensive understanding of the drug’s distribution landscape. Careful optimization of the electrophilic warhead’s selectivity and reactivity during lead optimization is paramount to minimize undesirable off-target interactions while maintaining potent on-target activity.
2. Metabolic Pathways: Conjugation and Adduct-Mediated Clearance Mechanisms
The metabolic fate of covalent drugs often involves unique clearance mechanisms, particularly conjugation pathways, due to their reactive electrophilic nature. A primary route of metabolic inactivation for many covalent drugs involves conjugation with endogenous nucleophiles, most notably glutathione (GSH) and cysteine. These pathways serve as crucial detoxification mechanisms, converting reactive electrophiles into more water-soluble and excretable metabolites.
For example, Futibatinib, an irreversible inhibitor of FGFR1-4 approved for FGFR2-rearranged cholangiocarcinoma, undergoes extensive metabolism via O-demethylation and glutathione conjugation. A detailed mass-balance study using 14C-futibatinib revealed that a major circulating metabolite was a cysteinylglycine conjugate, representing approximately 13% of the circulating drug-related material. Further in vitro hepatocyte studies identified additional GSH, cysteine, glucuronide, and sulfate metabolites, highlighting the complexity of its metabolic profile. The formation of these conjugates significantly contributes to the drug’s elimination from the body.
Beyond direct conjugation, covalent drugs can also be cleared through the breakdown and excretion of drug-protein adducts. Once a covalent drug binds to a protein, the entire adduct can be subject to proteolytic degradation and subsequent clearance. For highly abundant plasma proteins like albumin, the albumin-bound drug adducts themselves can represent a significant circulating pool that is slowly cleared, influencing the overall systemic exposure and elimination kinetics. This "adduct-mediated clearance" can significantly extend the drug’s presence in the body, even if the unbound drug is rapidly eliminated.
Advice for Developers: Early and thorough characterization of non-enzymatic and glutathione S-transferase (GST)-mediated reactivity is essential. Developers should quantify the electrophile’s reactivity, ranking potential candidates based on their propensity for GSH conjugation. Utilizing recombinant GST isoforms and human hepatocyte systems can help assess polymorphism-related risks, as genetic variations in GST enzymes can influence an individual’s capacity to detoxify reactive metabolites, potentially impacting drug safety and efficacy. Understanding these complex clearance routes is vital for predicting drug half-life, designing appropriate dosing regimens, and anticipating potential inter-individual variability in response.
3. Drug-Drug Interaction Risk: Time-Dependent CYP450 Inhibition
A significant concern with covalent drugs is their propensity for time-dependent inhibition (TDI) of cytochrome P450 (CYP450) enzymes, the primary enzymes responsible for drug metabolism in the liver. Unlike reversible inhibitors that compete for binding in real-time, TDIs form a stable complex with the enzyme, often leading to its irreversible inactivation or degradation. This means that the degree of inhibition increases over time and with repeated dosing, potentially leading to clinically significant drug-drug interactions (DDIs).
Studies have consistently shown that a substantial proportion of covalent drugs act as TDIs of at least one human CYP enzyme. A notable study by Moghaddam MF et al. (2014) evaluated ten diverse covalent drugs across multiple CYP isoforms and found that most exhibited time-dependent inhibition of at least one enzyme, underscoring the widespread nature of this risk. For instance, some covalent drugs might inactivate CYP3A4, a major metabolizing enzyme, leading to elevated plasma levels of co-administered drugs that are also substrates for CYP3A4, potentially causing toxicity. Conversely, if a co-administered drug is a prodrug requiring CYP activation, TDI could reduce its efficacy.
Advice for Developers: Early kinetic characterization of TDI, including determining the inhibition constant (KI) and the inactivation rate constant (kinact), is crucial. Mechanistic modeling, which integrates these kinetic parameters with drug concentration and dosing frequency, can effectively predict the clinical DDI risk. This proactive approach allows drug developers to either structurally optimize the compound to minimize TDI or implement specific dosing strategies (e.g., staggered administration, dose adjustments for co-medications) to mitigate potential interactions in clinical settings. Regulatory bodies increasingly require comprehensive TDI assessment as part of preclinical safety packages.
4. Decoupling PK/PD: A New Paradigm for Efficacy
Perhaps the most defining pharmacokinetic characteristic of covalent drugs is the fundamental decoupling of their plasma concentration from their pharmacodynamic (PD) effect. For traditional, reversible small-molecule drugs, maintaining a specific therapeutic concentration in the blood is typically essential to sustain the desired pharmacological effect. As the drug is metabolized and eliminated, its plasma concentration drops, and the effect diminishes.
Covalent drugs operate under a different paradigm. Once they form an irreversible bond with their target protein, they persistently occupy the active site. The duration of the pharmacological effect is then largely dictated not by the drug’s plasma half-life, but by the turnover rate of the target protein itself – specifically, the time required for the cell to synthesize new, active target protein. The drug’s presence in the systemic circulation becomes less critical after the initial target engagement.
A compelling illustration of this phenomenon comes from studies with CC-292, an irreversible BTK inhibitor. Despite plasma levels of CC-292 falling to near or even below the lower limit of quantification within 24 hours post-dose, target occupancy in preclinical and clinical studies remained high for up to 24 hours, declining only as new BTK protein was resynthesized. This stark contrast highlights that a drug’s plasma half-life, a cornerstone metric for reversible drugs, is a poor predictor of the duration of action for covalent inhibitors. Instead, the focus shifts to achieving sufficient initial exposure (Cmax-driven rapid engagement) to ensure robust target binding.
Advice for Developers: To effectively address the challenges presented by this PK/PD decoupling, researchers must adopt a target occupancy-driven development strategy. This involves directly modeling target occupancy using various in vivo techniques, such as mass spectrometry-based target engagement assays, biochemical assays in biopsy samples, or advanced imaging modalities like Positron Emission Tomography (PET) scans with radiolabeled probes. The primary goal becomes ensuring maximal target engagement in the therapeutic window, rather than simply maintaining plasma drug concentrations. Dosing regimens can then be optimized based on the turnover rate of the target protein, potentially allowing for less frequent dosing despite a short plasma half-life, thereby improving patient compliance and reducing systemic exposure to the unbound drug.
Industry Response and Future Outlook
The resurgence of covalent drugs has necessitated a significant adaptation within the pharmaceutical industry and among regulatory bodies. Drug developers are increasingly integrating specialized preclinical and clinical strategies to characterize these agents. This includes the routine use of advanced analytical techniques, such as high-resolution mass spectrometry for adduct identification, and sophisticated in vitro models to predict human metabolism and DDI risk. Computational chemistry and molecular modeling play a crucial role in the rational design of selective warheads and in predicting potential off-target interactions.
Regulatory agencies like the FDA and EMA have also evolved their guidance, emphasizing the need for comprehensive mechanistic understanding of covalent drug behavior. Submissions for covalent drugs now typically require extensive data on target engagement, off-target reactivity, TDI potential, and a clear demonstration of the PK/PD relationship that accounts for target turnover. This shift reflects a maturing understanding of their unique pharmacology.
The future of covalent drug development remains bright, with ongoing research exploring novel electrophilic warheads, new target classes, and innovative applications. Beyond traditional enzyme inhibition, covalent strategies are being explored in areas such as targeted protein degradation (e.g., PROTACs that can involve covalent linkers), gene editing tools, and antibody-drug conjugates. As technology continues to advance, development teams equipped with deep expertise in medicinal chemistry, molecular pharmacology, and quantitative PK/PD modeling will be best positioned to design, characterize, and bring these transformative therapies to patients efficiently and safely, addressing some of the most critical unmet medical needs in modern therapeutics. The careful, deliberate, and mechanistic understanding of covalent chemistry will continue to unlock new therapeutic frontiers, solidifying their role as indispensable tools in precision medicine.









Leave a Reply