
The pharmaceutical industry stands at a critical juncture, grappling with escalating costs and protracted timelines in the arduous journey of bringing life-saving drugs to market. Traditionally, the path from initial discovery to regulatory approval for a successful drug spans an average of 10 to 14 years, often extending even longer. This lengthy process is compounded by staggering financial demands, with the cost of developing a single new drug ranging from hundreds of millions to multiple billions of dollars. Alarmingly, this inflation-adjusted cost has roughly doubled every nine years, a phenomenon often referred to as "Eroom’s Law" (Moore’s Law spelled backward), highlighting a troubling inverse trend in productivity.
Estimates for R&D costs per approved drug exhibit considerable variation, influenced by methodologies and the inclusion of factors such as failed projects, cost of capital, and post-approval expenditures. Influential analyses, one leveraging confidential firm data, projected costs of approximately $1.4 billion out-of-pocket and $2.6 billion capitalized (in 2013 dollars). Conversely, a public-data analysis of FDA approvals placed the median capitalized research and development investment at $985.3 million and the mean at $1335.9 million in its base case. Regardless of the exact figures, the financial burden is immense and continually rising.
The Escalating Challenge of Traditional Drug Discovery
A significant driver of these costs and timelines is the notoriously high failure rate inherent in drug development. Only a minuscule fraction of compounds entering preclinical testing successfully advance to first-in-human studies. Furthermore, once a drug candidate enters clinical trials, only about 12% ultimately gain FDA approval. The financial repercussions of a late-stage trial failure, encompassing all sunk development costs, can range from $800 million to a staggering $1.4 billion.
Clinical development success rates are not uniform; they vary significantly by disease area and data set. A comprehensive benchmark study of programs conducted between 2011 and 2020 revealed an approximate 7.9% likelihood of approval from Phase I onwards across all therapeutic areas. This figure dips even lower for oncology, standing at roughly 5.3%. Other industry summaries have placed the overall success rate under 14%.

This economic reality has profound consequences, particularly for patients suffering from rare diseases. Despite affecting an estimated 3.5% to 5.9% of the global population—approximately 263 to 446 million people—a disheartening 95% of rare diseases in the U.S. currently lack FDA-approved treatment options. The high failure rates and associated financial risks mean that investment is predominantly channeled into developing drugs for more common "blockbuster" indications, where the potential return on investment is perceived to be greater. This creates a vicious cycle: the prohibitive cost of failure concentrates research efforts on diseases with larger patient populations, leaving millions with rare and complex conditions without viable therapies.
A New Era: The Rise of Digital Drug Design and AI
Breaking this cycle is the imperative driving the rapid adoption of digital drug design tools. These innovations, ranging from AI-driven target identification to generative molecular design, promise to compress timelines, substantially reduce costs, and broaden the spectrum of diseases that can be viably pursued. By streamlining various stages of discovery and development, digital tools and artificial intelligence (AI) are poised to revolutionize how new medicines are brought to patients.
From Serendipity to Supercomputers: A Chronology of Innovation
The history of drug discovery underscores the transformative power of technological advancement. Before the 1970s, the field was largely characterized by serendipitous observations and arduous, resource-intensive trial-and-error screening. Scientists possessed limited understanding of molecular mechanisms, relying heavily on phenotypic screening and empirical testing, a process that often spanned decades with no guarantee of success.
The paradigm began to shift in the 1970s with the emergence of computer-aided drug design (CADD). Initially a conceptual framework, CADD applications steadily grew throughout the 1980s and beyond. This approach harnessed computational techniques to analyze the structure and interactions of molecules with their biological targets. Early CADD methods were designed to assess the activity, toxicity, and bioavailability of potential drug candidates, offering a more systematic and predictive approach than traditional methods.

The subsequent decades witnessed continuous refinement and increasing accuracy in computational predictions. The 1980s saw the evolution of CADD into more specialized fields such as structure-based drug design (SBDD), which leverages the 3D structure of a target protein, and ligand-based drug design (LBDD), which focuses on the properties of known active compounds. The 2000s marked a significant acceleration, driven by advancements in structural biology, genomics, and bioinformatics. The exponential growth of resources like the Protein Data Bank (PDB), a global repository for 3D biological macromolecular structures, and other chemical libraries, made virtual screening a standard practice in drug discovery.
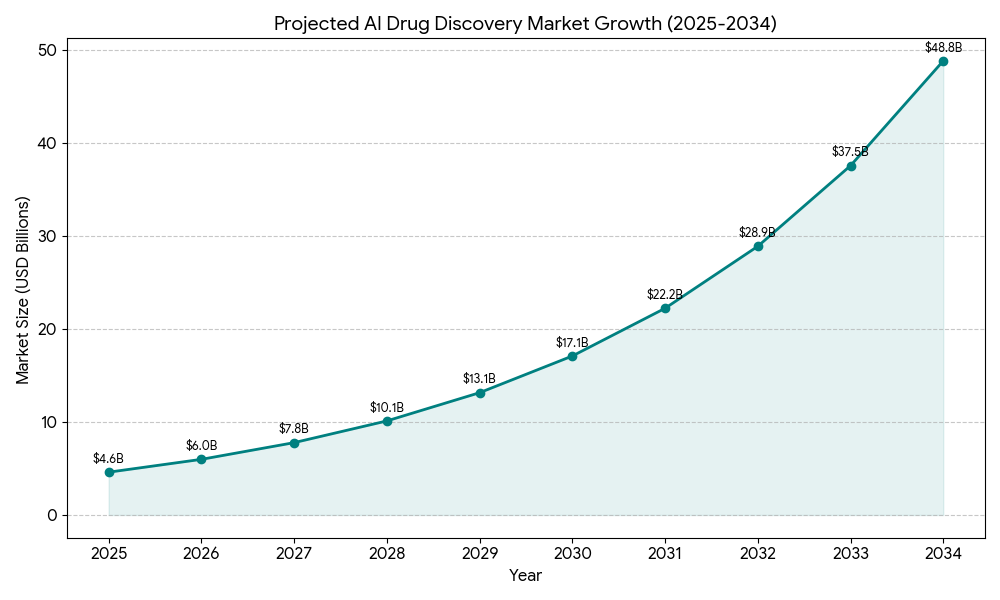
The 2020s have ushered in a truly revolutionary phase with the deep integration of AI and machine learning (ML) into CADD approaches. Deep learning models, capable of identifying intricate patterns in vast datasets, have enabled unprecedented predictive accuracy. Groundbreaking tools like AlphaFold, which can predict protein structures with astonishing precision, and other generative AI models have fundamentally transformed structure prediction and molecular design. This rapid acceleration is reflected in market projections: the AI drug discovery market is estimated to soar from $4.6 billion in 2025 to $49.5 billion by 2034, boasting a compound annual growth rate (CAGR) of 30%.
AI’s Transformative Impact Across the Discovery Pipeline
Today, "digital drug design" is an expansive and continually evolving domain. It encompasses a diverse toolkit, including sophisticated molecular simulations that model the dynamic interactions between compounds and biological targets; machine learning models meticulously trained on immense chemical libraries to predict optimal drug candidates; generative AI platforms that propose entirely novel molecular structures; and, increasingly, integrated lab automation platforms that seamlessly bridge computational predictions with physical experimental validation.
Adoption of these advanced technologies has dramatically accelerated in recent years. The first AI-designed drugs are now progressing through clinical trials, a testament to the technology’s maturity and potential. Companies such as Insilico Medicine, Recursion Pharmaceuticals, and Absci are at the forefront, showcasing end-to-end AI pipelines that demonstrate the feasibility of this new paradigm. The relentless pressure of rising R&D costs is further compelling pharmaceutical companies to embrace efficiency improvements, while recent successes, such as the GLP-1 receptor agonists, underscore the compelling return on investment potential of adopting these digital tools. Emerging platforms are increasingly integrating multiple tools, and AI-powered lab notebooks, like Sapio’s ELaiN, are moving beyond mere record-keeping to facilitate active, intelligent collaboration among researchers.
Molecular Dynamics Simulations: Beyond Static Structures
Molecular dynamics simulations have evolved significantly, moving beyond static structural analysis to capture the dynamic behavior and multiple conformations of molecules. Deep learning (DL) models are now adept at predicting a molecule’s properties and activity directly from its molecular structure. Convolutional neural networks (CNNs), traditionally used for image processing, can effectively analyze graphical representations of molecular structures to predict properties and activity. Recurrent neural networks (RNNs) leverage sequence data, capturing long-term dependencies within molecular sequences to enhance the accuracy of property predictions. Graph neural networks (GNNs) are particularly powerful, as they can directly process molecular graphs, modeling the complex relationships between atoms and bonds to predict diverse molecular properties.
Simplified molecular input line entry system (SMILES) is a widely adopted standard for drug characterization. This linear molecular representation allows SMILES strings to be directly processed as text, making them exceptionally useful in DL models for tasks such as inverse synthesis prediction, often employing sequence-to-sequence (seq-2-seq) methods. The ability of SMILES to generate multiple representations of the same molecule by varying atomic order provides a significant advantage for data augmentation, enhancing the robustness of DL models.
Furthermore, DL-based drug-target interaction (DTI) prediction models frequently utilize molecular fingerprints (MFPs) as input features. A molecular fingerprint is a bit string that compactly encodes the structural or pharmacological properties of a molecule. These fingerprints are invaluable in similarity searches and quantitative structure-activity relationship (QSAR) analysis, enabling rapid comparison and prediction of molecular behavior.
Drug Target Discovery: Unraveling Biological Complexity
Identifying and validating novel drug targets is a foundational step in drug discovery, and AI is proving instrumental here. AI algorithms can be seamlessly combined with systems biology approaches to uncover intricate correlations between multi-omics data (genomics, proteomics, metabolomics) and patient clinical health information. Natural language processing (NLP) methods are deployed to extract and analyze unstructured data from vast repositories of scientific literature, patents, and clinical reports, thereby identifying potentially relevant biological pathways and disease mechanisms that might otherwise remain hidden.
The inherent complexity of biological systems presents formidable challenges in constructing predictive models for disease classification. Traditional models often struggle to provide stable pathways for specific phenotypes or reliable biomarkers of disease. However, advanced Bayesian AI analysis offers a solution, integrating diverse molecular profiles and multi-omics data with clinical health information to construct causal inference networks. These networks are powerful tools for identifying robust targets and reliable biomarkers, paving the way for more precise therapeutic interventions.

Target Structure Confirmation: Accelerating Visualization
Target structure confirmation, another critical process, has also been significantly enhanced by digital tools. Databases like the Potential Drug Target Database (PDTD), released in 2008, house over 1,100 3D protein structures covering 830 drug targets. This resource provides protein and active site structures in various formats, alongside related disease information, biological functions, and associated regulating and signaling pathways, vastly accelerating the initial stages of structural analysis.
Traditional methods for protein structural resolution, such as nuclear magnetic resonance (NMR), X-ray crystallography, and cryo-electron microscopy, offer invaluable insights into protein structures and drug receptors. However, these techniques are capital-intensive, requiring expensive equipment, and their timelines can range from weeks to years, contingent on sample availability and protein complexity. AI-driven solutions, particularly in protein folding prediction like AlphaFold, are dramatically reducing the time and cost associated with obtaining accurate 3D protein structures, democratizing access to this crucial information.
Virtual Screening: Precision at Scale
Virtual screening (VS) methods are pivotal in rapidly sifting through vast chemical libraries to identify promising drug candidates. Structure-based virtual screening (SBVS), or target-based VS, is a CADD method that predicts the interaction between a target protein and a library of compounds using the target’s 3D structure. It ranks compounds based on their predicted affinity for the target’s receptor binding site. Molecular docking, a key SBVS technique, examines the geometric compatibility between ligands and targets. While widely used, the intricate nature of ligand-receptor binding interactions can lead to difficulties in accurately predicting binding sites and classifying compounds, often resulting in high false-positive and false-negative rates. The accuracy of SBVS is heavily reliant on the sophistication of the search algorithm and scoring function.
Ligand-based virtual screening (LBVS), conversely, identifies bioactive compounds by analyzing the chemical and structural features of known ligands. Traditional LBVS methods include molecular similarity searches and quantitative structure-activity relationship (QSAR) modeling. Recent advances in AI have introduced machine learning (ML) and deep learning (DL) techniques to LBVS, enhancing molecular representation, optimizing similarity searches, and refining the accuracy of QSAR models. AI-powered frameworks have also addressed challenges posed by data scarcity by incorporating multi-task learning, transfer learning, and graph-based neural networks.
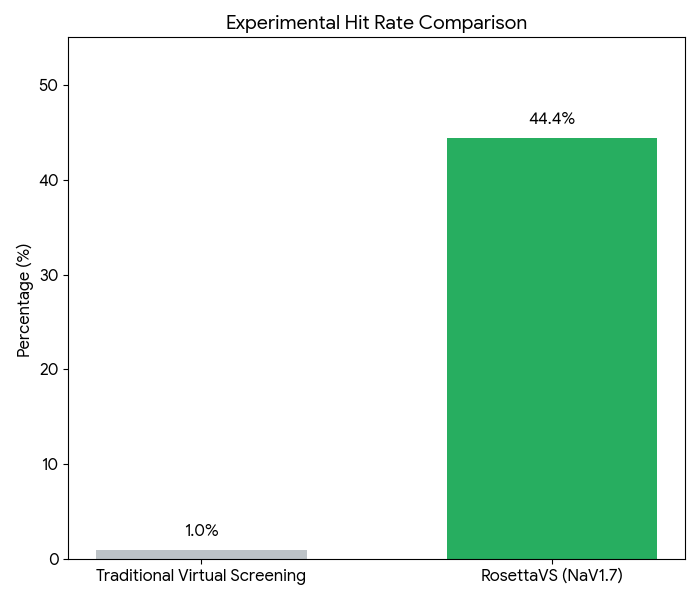
AI-based VS models often utilize molecular descriptors derived from physicochemical properties or fingerprints to construct regression or classification models of activity, offering greater flexibility than traditional LBVS. DL-based molecular docking leverages AI algorithms to accelerate docking processes and improve the accuracy of pose predictions. Platforms such as BioNeMo, DiffDock, and Rosetta VS integrate DL with other advanced methods to screen molecules with unprecedented speed and precision. A compelling case study involving RosettaVS on NaV1.7 demonstrated the power of this approach: the team employed active learning to triage a multi-billion compound space, docked 4.5 million compounds, synthesized 9 candidates, and observed 4 micromolar binders, achieving an impressive 44.4% hit rate among the synthesized compounds. This illustrates a profound shift in efficiency.
Synthetic Routes: Charting the Path to Production
Identifying an efficient and cost-effective synthetic route is a critical, often labor-intensive, aspect of drug development. Computer-aided synthetic planning (CASP) assists chemists by identifying optimal synthesis pathways, predicting selectivity and byproducts, and suggesting ideal reaction conditions. CASP has evolved from systems based on manually coded reaction rules and templates to sophisticated AI-assisted synthesis planning. Modern AI algorithms can recommend synthetic routes for a wide range of reactions, even without predefined templates, utilizing molecular representations like fingerprints, graphs, or SMILES strings.
While rule-based approaches, often termed "template methods," rely on coded rules and heuristics extracted from extensive reaction databases and literature, they are limited by their inability to scale with the exponential growth of chemical literature and the inherent incompleteness of human-curated knowledge bases. Inverse synthesis software like Synthia addresses these limitations by integrating computational methods with an expert-coded rule base, accumulated over 21 years and comprising more than 115,000 rules, alongside a catalog of over 12 million commercially available starting materials. This powerful combination accelerates pathway design, using scoring functions and dynamic planning algorithms to construct and propose optimal synthetic routes.
Furthermore, template-free approaches, drawing inspiration from natural language processing (NLP), frame synthetic prediction as a sequence-to-sequence (seq-2-seq) mapping problem. By representing chemical reactions as "sentences" using SMILES strings, this method treats the process as a chemical language translation, enabling the prediction of novel synthetic pathways without explicit rules.
Revolutionizing Clinical Trials: Mitigating Risk and Accelerating Progress
Clinical trials represent the most expensive and time-consuming phase of drug development, with failure rates from Phase I to approval reaching as high as 95% in certain disease areas, including oncology. Given these staggering figures, AI offers a potential lifeline. In vivo studies, primarily clinical trials, account for the majority of new entity development costs; one estimate attributes approximately $1.4 billion of a $2.3 billion total to clinical trials. This highlights that while computational improvements in early drug discovery are crucial, their direct impact on the overall cost of bringing a drug to market is limited without addressing clinical trial efficiency.
Models capable of predicting late-stage clinical trial outcomes hold immense potential for improving success rates. Concepts like the Virtual Physiological Human (VPH), proposed in a 2005 white paper, envision using virtual twin technology to support clinical decision-making and predict safety and efficacy. However, accurately modeling the complex biology of the human organism remains a significant challenge, and the accuracy of in silico clinical trial predictions requires further rigorous study and validation.
Consequently, current AI technologies in this domain primarily focus on integrating patient genetic data, electronic health records (EHRs), medical literature, and clinical trial databases to predict trial success. Other AI models are designed to optimize trial design, assist with precise patient matching and recruitment, and enhance monitoring of patient adherence during trials.
Toxicity is a frequent cause of clinical trial failures, and computational models are increasingly adept at predicting it. PrOCTOR, a tree-based ensemble ML model, combines chemical features of drugs with target-based features to distinguish between approved drugs and those that failed due to toxicity. Drugs predicted as toxic by PrOCTOR were indeed found to have significantly more frequent reports of serious adverse events than those predicted as safe. Insilico Medicine has developed a deep neural network (DNN) based on pathway analysis to predict drug side effects by analyzing transcriptional changes induced by drugs, forecasting clinical trial outcomes for 46 different side effects. Their inClinico platform provides forecasts of clinical trial probability of success, identifies weak points in trial design, and suggests optimizations. Other companies like Unlearn.ai, Saama, and Phesi offer similar services, leveraging AI to de-risk and optimize clinical development.
Natural language processing (NLP) techniques are also transforming patient recruitment. NLP-based trial matching systems, notably demonstrated by IBM Watson teams, extract information from electronic medical records (EMRs) to create detailed clinical profiles for patients. These profiles are then compared against the eligibility requirements of available clinical trials, significantly speeding up the matching process and ensuring suitable patient populations.
AI is also being deployed to enhance patient adherence tracking in clinical trials. Historically, trials relied on imprecise methods such as pill counts and self-reported data, both prone to errors and manipulation. Now, AI improves accuracy. AbbVie, for instance, implemented an AI-powered facial and image recognition system through the AiCure mobile program, requiring patients to upload video evidence of swallowing pills. The AI system verifies the correct person took the prescribed medication. A study involving schizophrenia patients showed a remarkable difference: patients monitored with AI achieved an adherence rate of 89.7%, compared to 71.9% for those monitored with modified directly observed therapy (mDOT) over 24 weeks—a 17.9% improvement. This is particularly significant given that typical adherence rates for schizophrenia patients hover around 50%.

Conclusion: Navigating the Future of Drug Discovery
Digital and simulation tools have steadily improved the drug discovery process since the 1980s, evolving from early CADD methods and the establishment of resources like the PDB to the profound integration of AI in the 2020s. These advancements are undeniably instrumental in bringing new, more effective medicines to market faster and more efficiently.
However, the rapid emergence and integration of AI also present a unique set of challenges. Trust in generative AI models, for instance, remains a critical issue due to the potential for "hallucinations" or misinformation. Many AI models are often regarded as "black boxes," providing accurate predictions without transparently revealing their decision-making processes. In a high-stakes industry like healthcare, transparency, alongside accuracy, is paramount for regulatory approval and physician acceptance.
Data scarcity poses another significant hurdle, particularly in rare disease areas. AI models often require training on millions, or even billions, of high-quality data samples to achieve optimal performance, a volume simply unavailable for many rare conditions. Even with seemingly clean data, inherent biases can inadvertently creep into models from training datasets, leading to skewed outcomes related to race, gender, and other demographic factors. Addressing these biases is crucial for equitable healthcare outcomes.
Furthermore, the environmental footprint of AI models cannot be ignored. The operation of these computationally intensive models consumes vast amounts of water and electricity. As the pharmaceutical and healthcare industry already faces scrutiny for its CO2 emissions and waste products, the continued integration of AI presents an environmental challenge that necessitates sustainable development and deployment strategies.
Despite these challenges, the immense promise of AI to drastically decrease the timelines and costs associated with drug discovery is undeniable. By accelerating every stage from target identification to clinical trials, AI holds the potential to deliver life-saving drugs to patients faster and more affordably. While obstacles remain, the transformative impact of these methods on the field and the entire process of drug discovery and development is profound and irreversible, heralding a new era for medicine.









Leave a Reply