
The landscape of drug discovery and development is witnessing a significant resurgence in covalent inhibitors, a class of therapeutics that form a permanent bond with their biological targets. From the early, serendipitous discoveries of aspirin and penicillin to the sophisticated, rationally designed targeted covalent inhibitors (TCIs) of today, these drugs have proven transformative, offering novel approaches to treat previously "undruggable" diseases. However, their unique mechanism of action introduces distinct pharmacokinetic (PK) and pharmacodynamic (PD) properties that diverge significantly from conventional reversible small-molecule drugs, posing specific challenges for development teams and regulatory bodies. Successfully navigating these complexities is crucial for realizing the full therapeutic potential of this innovative drug class.
A Historical Perspective: From Serendipity to Strategic Design
Covalent drugs have a long and impactful history, though their mechanisms were often understood decades after their initial clinical success. Aspirin, approved in 1899, stands as a foundational example, its anti-inflammatory and analgesic effects later attributed to the irreversible acetylation of cyclooxygenase (COX) enzymes. Penicillin, a monumental breakthrough in infectious disease treatment, similarly operates by forming a covalent bond with bacterial transpeptidase enzymes, inhibiting cell wall synthesis. These early successes, while revolutionary, were largely accidental, with the intricate molecular details of their covalent interactions remaining elusive for many years. It took 70 years for aspirin’s COX inhibition mechanism to be fully elucidated, and over 50 years for penicillin’s covalent binding to bacterial enzymes to be understood.
However, the mid-20th century saw a pronounced shift away from covalent drug development. The primary concern revolved around the indiscriminate reactivity of electrophilic compounds, which were often metabolized into highly reactive species capable of binding non-selectively to various biomolecules, including proteins, lipids, and DNA. This non-specific binding could lead to cellular damage, off-target toxicities, and, in some cases, carcinogenicity. High-profile instances of toxicity associated with compounds like acetaminophen (hepatic toxicity), bromobenzene, and urushiol (a component of poison ivy) solidified a prevailing view that electrophiles were "no-go" zones for drug development. Consequently, medicinal chemistry efforts largely gravitated towards the design of reversible, non-covalent inhibitors, which were perceived as inherently safer due to their transient interactions with targets. This era effectively sidelined covalent drug research for several decades, limiting innovation in this promising area.
The Renaissance of Covalent Drug Development
The past decade has heralded a remarkable renaissance in covalent drug development, driven by a profound shift in design philosophy and a deeper understanding of electrophile chemistry. Modern covalent drugs employ "soft electrophiles" such as acrylamide, nitrile, and vinyl sulfone warheads. These groups are engineered to possess attenuated reactivity, allowing for selective engagement with specific nucleophilic residues (most commonly cysteine thiols) within a target protein’s active site, while minimizing promiscuous off-target binding. This rational design approach, central to the concept of Targeted Covalent Inhibitors (TCIs), has fundamentally dispelled the earlier dogma that all electrophiles are inherently unsafe and promiscuous.
The importance of carefully tuning electrophilicity cannot be overstated. A compelling illustration of this principle lies in the comparison between sucralose, a widely used artificial sweetener, and sulfur mustard, a chemical weapon. Both contain alkyl chloride groups. Yet, sucralose is safe for chronic consumption, whereas sulfur mustard causes severe alkylation damage due due to its high reactivity. This stark contrast underscores that it is not merely the presence of an electrophilic moiety but its carefully controlled reactivity and selectivity that dictates safety and efficacy.
The success of TCIs, exemplified by drugs like ibrutinib (a Bruton’s tyrosine kinase, BTK, inhibitor for hematologic malignancies), osimertinib (an EGFR inhibitor for non-small cell lung cancer), and futibatinib (an FGFR inhibitor for cholangiocarcinoma), has catalyzed a boom in the field. To date, over 110 covalent drugs have received regulatory approval across diverse therapeutic areas, including infectious diseases (e.g., penicillin), gastrointestinal disorders (e.g., omeprazole), hepatitis C (e.g., telaprevir), and oncology. This resurgence is further supported by the increasing sophistication of analytical tools and computational methods, enabling researchers to precisely evaluate parameters such as reaction kinetics (e.g., kinact/Ki), glutathione (GSH) conjugation potential, and time-dependent inhibition (TDI) risk of cytochrome P450 enzymes. Despite these advancements, the unique binding mechanism of covalent drugs continues to present distinct pharmacokinetic challenges that demand specialized strategies throughout the drug development pipeline.
Navigating the Unique Pharmacokinetic Landscape
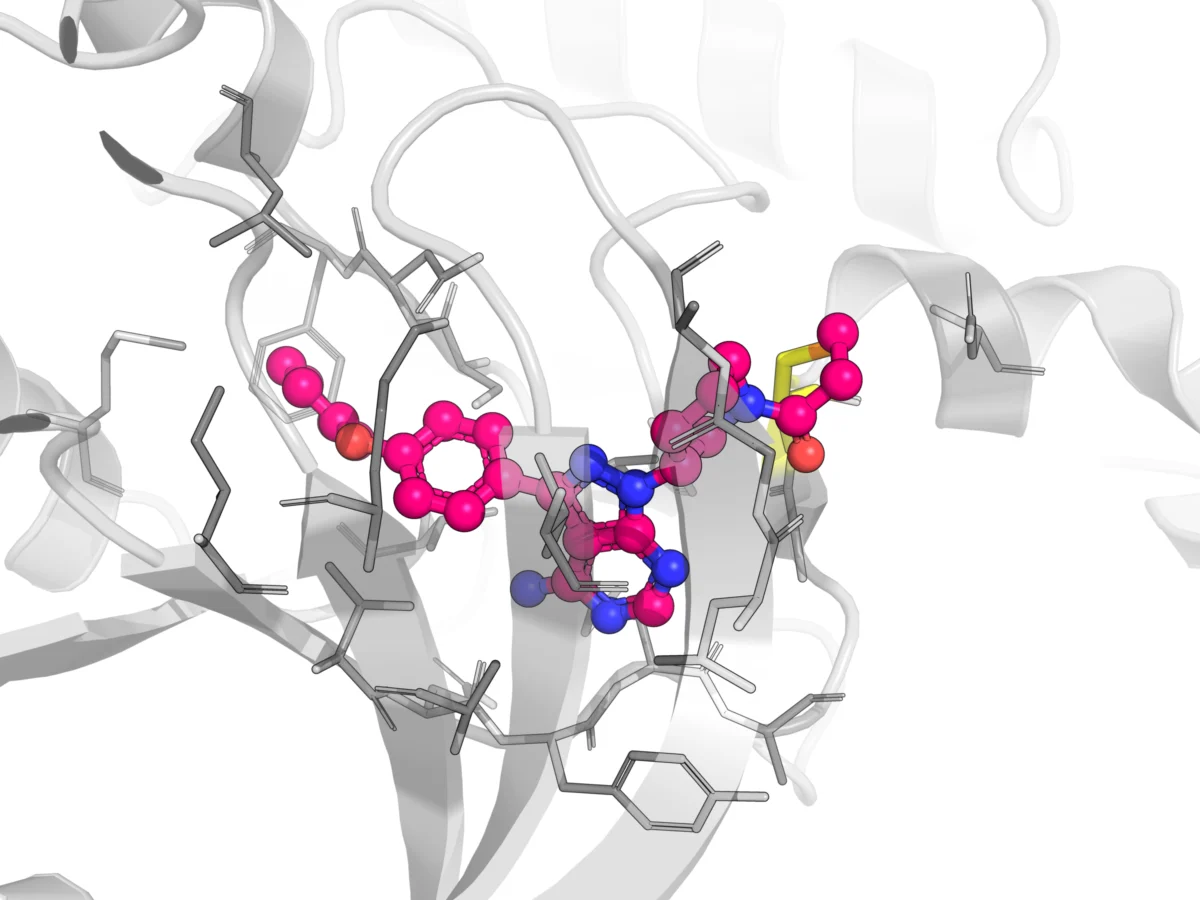
Covalent drugs, by their very nature, establish an irreversible or quasi-irreversible bond with their target. This fundamental characteristic leads to pharmacokinetic properties that differ significantly from those of conventional, reversibly binding small-molecule drugs. Key considerations for developers include the complexities of protein binding beyond the primary target, novel clearance mechanisms involving protein-adducts, the propensity for time-dependent inhibition of metabolizing enzymes, and a distinct relationship between plasma drug concentration and pharmacological effect.
I. Protein Binding Beyond the Target Protein
While covalent binding to the intended therapeutic target is paramount for efficacy, covalent drugs also possess the potential to bind covalently to other proteins throughout the body. These off-target interactions can profoundly influence a drug’s distribution, metabolism, and potential for toxicity. For instance, covalent drugs can form stable adducts with highly abundant proteins like human serum albumin (HSA) in the plasma. This binding can significantly reduce the concentration of unbound, pharmacologically active drug, thereby impacting its distribution to target tissues and potentially influencing its clearance. Similarly, covalent binding to tissue proteins, particularly within highly metabolic organs such as the liver, can affect local drug retention, alter metabolic pathways, and contribute to organ-specific toxicities.
A critical challenge for developers is to meticulously differentiate between therapeutically relevant on-target engagement and undesirable off-target reactivity. High levels of off-target protein binding do not inherently correlate with adverse effects; rather, the selectivity of these interactions is key. For example, some covalent drugs might form adducts with plasma proteins that act as a "depot," slowly releasing the drug or its metabolites without causing harm. Conversely, binding to critical enzymes or structural proteins in non-target tissues could lead to toxicity. Therefore, meticulous optimization of both the selectivity and the reactivity of the electrophilic warhead during the lead optimization phase is essential to minimize off-target effects and enhance the therapeutic index. Advanced proteomic techniques and in vitro assays designed to assess broad protein reactivity can provide crucial insights early in development.
II. Conjugation and Protein-Adduct-Mediated Clearance Mechanisms
The metabolic fate of covalent drugs often involves unique clearance pathways, primarily through conjugation reactions. Glutathione (GSH) conjugation and cysteine adduct formation represent major routes of metabolic inactivation for many electrophilic compounds, including covalent drugs. These pathways are crucial detoxification mechanisms, converting reactive electrophiles into more water-soluble and excretable metabolites.
Futibatinib, an irreversible FGFR1-4 inhibitor approved for FGFR2-rearranged cholangiocarcinoma, provides a pertinent example. Its primary metabolic routes involve O-demethylation and extensive glutathione conjugation. A mass-balance study using 14C-futibatinib revealed that a major circulating metabolite was a cysteinylglycine conjugate, accounting for approximately 13% of the circulating radioactivity. Further in vitro hepatocyte studies demonstrated the formation of additional GSH, cysteine, glucuronide, and sulfate metabolites, underscoring the complexity of its metabolic profile.
For developers, it is imperative to quantify both non-enzymatic and glutathione S-transferase (GST)-mediated reactivity early in the discovery process. This allows for the ranking of electrophile reactivity and provides insight into potential metabolic liabilities. Utilizing recombinant GST isoforms and hepatocyte systems is critical to assess polymorphism-related risks, as genetic variations in GST enzymes can significantly alter a patient’s capacity to detoxify certain electrophiles, leading to variable drug exposure and toxicity. Furthermore, covalent drugs can also be cleared through the breakdown and subsequent elimination of drug-protein adducts, such as those formed with albumin. This "adduct-mediated clearance" can represent a significant portion of the total drug excretion, requiring specialized analytical methods to track and quantify these complex species. Understanding these mechanisms is vital for accurate PK modeling and predicting human exposure.
III. Time-Dependent Inhibition (TDI) of CYP450 Enzymes
A significant concern in drug development is the potential for drug-drug interactions (DDIs), often mediated by the inhibition or induction of cytochrome P450 (CYP450) enzymes, the primary enzymes responsible for drug metabolism. For covalent drugs, the risk of time-dependent inhibition (TDI) of CYP450 enzymes is particularly pronounced. TDI occurs when a drug or its metabolite irreversibly inactivates a CYP enzyme, often through covalent binding to the enzyme’s active site. This leads to a progressive decrease in enzyme activity over time, which can result in elevated plasma concentrations of co-administered drugs that are substrates for the inhibited CYP enzyme.
Studies have consistently shown that a considerable proportion of covalent drugs act as TDIs of at least one human CYP enzyme. For instance, a seminal study by Moghaddam MF et al. (2014) evaluated ten diverse covalent drugs across multiple CYP isoforms and found that most exhibited time-dependent inhibition of at least one enzyme. This inherent characteristic suggests that many covalent drugs carry an elevated risk of clinically significant DDIs.
To mitigate this risk, early and comprehensive kinetic characterization is indispensable. This includes determining parameters such as the inhibition constant (KI) and the maximal inactivation rate (kinact) for relevant CYP isoforms. Mechanistic modeling, integrating these kinetic parameters, can help predict the likelihood and magnitude of clinical DDI risk. Such data are crucial for guiding structural optimization strategies to reduce TDI potential, or for informing appropriate dosing adjustments and labeling recommendations if a TDI risk cannot be entirely eliminated. Proactive assessment allows for informed decisions that can prevent adverse drug events in patients.
IV. The Decoupling of PK/PD Relationship
Perhaps the most distinctive and challenging pharmacokinetic characteristic of covalent drugs is the decoupling of their plasma concentration-time profile (PK) from their pharmacological effect-time profile (PD). For traditional, reversible small-molecule drugs, a continuous presence of the drug at or above a specific concentration in the blood (and thus at the target site) is generally required to sustain the desired pharmacological effect. Plasma half-life often serves as a good predictor of dosing frequency.
However, once a covalent drug forms an irreversible bond with its target protein, it persistently occupies the active site. The duration of the pharmacological effect is then largely determined not by the drug’s plasma half-life, but by the turnover rate of the target protein itself – specifically, the time required for the cell to synthesize new, functional target protein molecules. The drug molecule, having performed its function, may be cleared from the plasma rapidly, yet its effect persists.
This phenomenon is vividly illustrated by irreversible BTK inhibitors like ibrutinib and CC-292. In studies with CC-292, plasma levels often fell to near or even below the lower limit of quantification within 24 hours post-dose. Despite this rapid plasma clearance, target occupancy in patients remained high for up to 24 hours and only began to decline as new BTK protein was synthesized by the cells. This critical decoupling implies that optimizing plasma exposure, as is typical for reversible drugs, is less critical than achieving rapid and sufficient initial target engagement. A high Cmax (maximum plasma concentration) that ensures complete target saturation shortly after dosing becomes paramount.
For developers, this necessitates a paradigm shift in PK/PD modeling. Instead of focusing solely on plasma drug concentrations, research should prioritize directly modeling target occupancy and correlating it with pharmacological endpoints. Dosing strategies should be tailored to achieve rapid, robust target binding, with subsequent dosing intervals dictated by the target protein’s resynthesis rate rather than the drug’s systemic clearance. This Cmax-driven rapid engagement approach is fundamental to designing effective and safe covalent drug regimens.
Broader Impact and Future Directions
The unique pharmacokinetic profile of covalent drugs has significant implications across the entire drug development ecosystem, from early discovery to regulatory approval and clinical practice. Regulatory bodies, such as the FDA and EMA, are increasingly adapting their guidelines to accommodate the specific data requirements for covalent inhibitors, demanding a thorough mechanistic understanding of safety, selectivity, and exposure. This includes detailed characterization of target engagement, off-target reactivity, and the implications of protein-adduct formation.
In the industry, there is a growing recognition of the need for specialized expertise in designing, characterizing, and modeling these complex mechanisms. Contract research organizations (CROs) with robust capabilities in covalent drug PK/PD are becoming invaluable partners, offering advanced analytical techniques for adduct detection, in vitro metabolism studies, and sophisticated PK/PD modeling. The continuous investment in computational chemistry and in silico modeling tools is also accelerating, allowing for more precise predictions of electrophile reactivity and selectivity, thereby streamlining the design process and reducing experimental burden.
For patients, the resurgence of covalent drugs holds immense promise. They offer therapeutic solutions for diseases previously deemed "undruggable" due to the transient nature of reversible inhibitors or the lack of suitable binding pockets. By providing sustained target inhibition independent of continuous plasma exposure, covalent drugs can potentially lead to improved efficacy, reduced dosing frequency for some indications, and better patient compliance. This aligns perfectly with the principles of precision medicine, where therapies are tailored to individual patient characteristics and disease biology.
In conclusion, covalent drugs represent a powerful and expanding frontier in modern therapeutics. To fully capitalize on their transformative potential, developers must embrace a nuanced and sophisticated understanding of their unique pharmacokinetic properties. Moving forward, successful covalent drug programs will be those that demonstrate deliberate and selective chemistry, backed by a comprehensive mechanistic understanding of safety, target engagement, and exposure. As technology continues to advance and our understanding deepens, development teams equipped with this specialized expertise will be best positioned to bring these innovative and life-changing therapies to patients efficiently and safely, pushing the boundaries of what is possible in medicine.









Leave a Reply