
The landscape of pharmaceutical innovation has long been characterized by arduous timelines, exorbitant costs, and a high probability of failure, creating significant hurdles in bringing life-saving treatments to patients. Traditional drug discovery pipelines typically span 10 to 14 years, sometimes longer, from initial discovery to market approval. This protracted process is compounded by escalating financial burdens; the inflation-adjusted cost of developing a drug has roughly doubled every nine years, a phenomenon often referred to as "Eroom’s Law" (Moore’s Law spelled backward), highlighting the decreasing efficiency of pharmaceutical R&D despite technological advances.
Published estimates for the R&D cost per approved drug vary, reflecting different methodologies and inclusions (e.g., failures, cost of capital, post-approval work). Influential analyses have pegged these costs anywhere from approximately $1.4 billion out-of-pocket and $2.6 billion capitalized (in 2013 dollars) to a median capitalized investment of $985.3 million and a mean of $1335.9 million for new drugs in base case analyses using public data. These figures underscore the immense financial risk inherent in drug development.
The journey from compound to cure is fraught with uncertainty. Only a minuscule fraction of compounds entering preclinical testing ever progress to human studies. Furthermore, only about 12% of drugs that successfully reach clinical trials will ultimately gain FDA approval. The financial fallout from a late-stage trial failure, encompassing sunk development costs, can range from $800 million to $1.4 billion. Clinical development success rates fluctuate based on disease area and dataset, with a large benchmark of programs from 2011–2020 estimating an overall 7.9% likelihood of approval from Phase I. Oncology, a notoriously challenging field, saw an even lower success rate of approximately 5.3% during this period, while other industry summaries place the overall figure under 14%.
This economic reality has fostered a vicious cycle: the monumental cost of failure compels investment to concentrate on "blockbuster" indications—diseases affecting large populations that promise substantial financial returns. Consequently, patients suffering from rare and complex diseases, despite representing 3.5% to 5.9% of the world’s population (263 to 446 million people), are often left without treatment options. A staggering 95% of rare diseases lack FDA-approved therapies in the U.S. This critical unmet medical need highlights a profound market failure, driven by the perceived lack of return on investment for less common conditions. However, a transformative shift is underway. Digital drug design tools, powered by artificial intelligence (AI) and machine learning (ML), are beginning to disrupt this cycle by dramatically compressing timelines, reducing costs, and expanding the range of diseases that are economically viable targets for therapeutic intervention.

The Evolution of Digital Drug Design: A Historical Perspective
The application of computational methods in drug discovery is not a sudden phenomenon but the culmination of decades of scientific and technological advancement. Before the 1970s, drug discovery was largely an empirical process, reliant on serendipitous observations (like the discovery of penicillin) and laborious trial-and-error screening of vast chemical libraries. This era, characterized by a rudimentary understanding of molecular mechanisms, often took decades and consumed substantial resources with no guarantee of success.
The 1970s marked the conceptual birth of Computer-Aided Drug Design (CADD), which gradually gained traction through the 1980s and beyond. CADD leveraged nascent computational techniques to analyze the structure and interactions of molecules with biological drug targets. Early applications focused on molecular modeling, allowing scientists to visualize and predict the activity, toxicity, and bioavailability of potential drug candidates in a virtual environment. This marked a crucial departure from purely experimental screening, offering a more rational and directed approach.
The subsequent decades witnessed continuous refinement and increased accuracy in CADD predictions. The 1980s saw the emergence of more sophisticated approaches such as structure-based drug design (SBDD), which utilizes the 3D structure of the target protein, and ligand-based drug design (LBDD), which relies on the properties of known active compounds. The 2000s were pivotal, as advancements in structural biology (e.g., X-ray crystallography, Nuclear Magnetic Resonance spectroscopy), genomics, and bioinformatics provided an unprecedented wealth of data. Resources like the Protein Data Bank (PDB) and ever-expanding chemical libraries became indispensable, making virtual screening a standard practice. These developments allowed researchers to screen millions of compounds computationally, significantly narrowing down the pool of candidates for experimental validation.
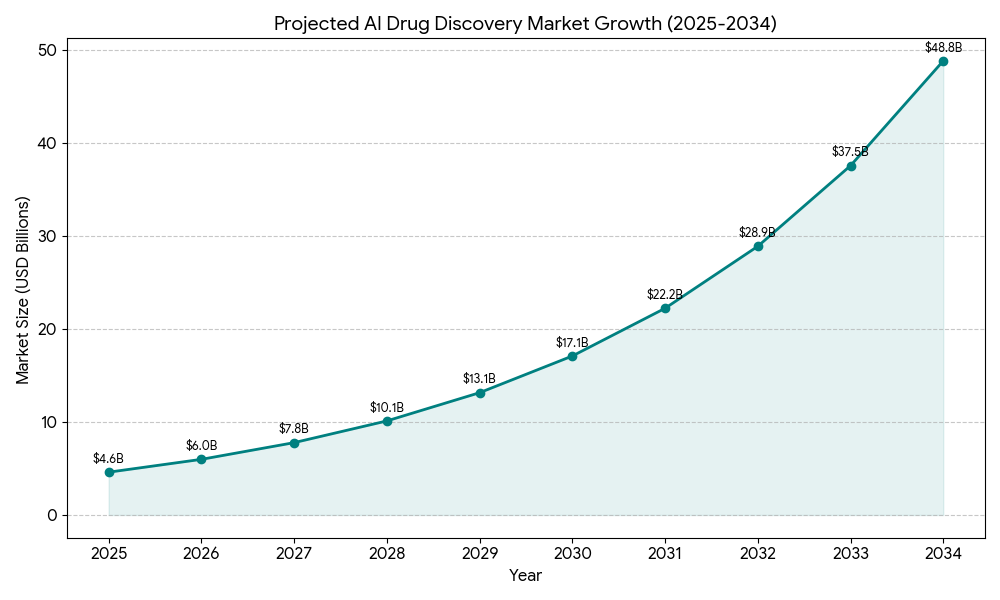
The 2020s ushered in the era of deep integration of AI and ML into CADD. Deep learning models, capable of identifying intricate patterns in massive datasets, enabled unprecedented predictive accuracy in areas like drug-target interaction and compound property prediction. Groundbreaking innovations such as AlphaFold, developed by DeepMind, revolutionized protein structure prediction, enabling scientists to accurately predict the 3D shapes of proteins from their amino acid sequences in minutes, a task that previously took years of laborious experimental work. Other generative AI tools further transformed molecular design, allowing for the de novo creation of entirely novel molecular structures optimized for specific therapeutic goals. This rapid acceleration is reflected in market projections, with the AI drug discovery market estimated to surge from $4.6 billion in 2025 to an astounding $49.5 billion by 2034, representing a Compound Annual Growth Rate (CAGR) of 30%. This growth underscores the industry’s widespread recognition of AI’s transformative potential.
The Present Moment: A Digital Renaissance in Pharma
Today, "digital drug design" is an expansive and rapidly evolving field, encompassing a diverse suite of tools that are fundamentally reshaping pharmaceutical R&D. These include sophisticated molecular simulations that model the dynamic interactions between compounds and biological targets, machine learning models meticulously trained on vast chemical libraries to predict the efficacy and safety of drug candidates, and generative AI platforms capable of proposing entirely novel molecular structures tailored to specific therapeutic profiles. Crucially, these computational predictions are increasingly linked to advanced lab automation platforms, creating integrated "wet lab-dry lab" ecosystems that streamline the entire discovery process.

The adoption of these technologies has accelerated dramatically in recent years, driven by the persistent pressures of rising R&D costs and the compelling evidence of return on investment. Several AI-designed drugs are now progressing through clinical trials, a testament to the maturity and efficacy of these new paradigms. Companies like Insilico Medicine, Recursion, and Absci are at the forefront, demonstrating end-to-end AI pipelines that integrate various digital tools from target identification to clinical candidate selection. The success of therapies like GLP-1 agonists, which leverage a deep understanding of biological pathways, further exemplifies the potential for digital tools to identify and optimize highly effective treatments.
Beyond individual tools, the trend is towards integrated platforms. AI-powered lab notebooks, such as Sapio’s ELaiN, are transcending mere record-keeping to become active collaborators in the research process, offering real-time data analysis, predictive insights, and automated experimental design. This holistic approach ensures that data generated in the lab feeds directly into computational models, and vice-versa, creating a continuous feedback loop that optimizes discovery.
Key Pillars of AI-Driven Drug Discovery
The impact of AI and digital tools is being felt across every stage of the drug discovery and development pipeline:
Molecular Dynamics Simulations
Modern molecular dynamics simulations have moved far beyond static structures, embracing the dynamic nature of biological molecules. Deep learning (DL) models are now adept at predicting a molecule’s properties and activity based on its intricate structure. Convolutional neural networks (CNNs) excel at processing molecular structure diagrams as image data, identifying spatial patterns crucial for predicting properties. Recurrent neural networks (RNNs) are utilized for sequence data, effectively capturing long-term dependencies within molecular sequences to enhance prediction accuracy. Graph neural networks (GNNs), a particularly powerful innovation, process molecular graphs to model the complex relationships between atoms and molecules, offering highly accurate property predictions.
The Simplified Molecular Input Line Entry System (SMILES) provides a linear molecular representation, allowing SMILES strings to be processed directly as text. This makes it invaluable in DL models for tasks such as inverse synthesis prediction using sequence-to-sequence (seq-2-seq) methods, akin to language translation. SMILES’ ability to generate multiple representations of the same molecule by altering atomic order also provides a significant advantage for data augmentation, improving model robustness. Furthermore, DL-based drug-target interaction (DTI) prediction models frequently use molecular fingerprints (MFPs) as input features. MFPs are bit strings that encode the structural or pharmacological properties of a molecule, widely used in similarity searches and quantitative structure-activity relationship (QSAR) analysis to rapidly assess potential compound activity.
Drug Target Discovery
Identifying the right biological target is the foundational step in drug discovery. AI algorithms, combined with systems biology approaches, can sift through vast multi-omics data (genomics, proteomics, metabolomics, etc.) and correlate it with patient clinical health information to uncover novel disease mechanisms and potential targets. Natural Language Processing (NLP) methods play a crucial role by extracting and analyzing unstructured data from scientific literature, patents, and clinical reports, revealing potentially relevant pathways and mechanisms that might otherwise remain buried.
However, the inherent complexity of biological systems presents significant challenges in constructing stable models for disease classification and identifying reliable biomarkers. To address this, Bayesian AI analysis has emerged as a powerful tool. By integrating molecular profiles and multi-omics data with clinical health information, Bayesian networks can construct causal inference networks, providing a more robust framework for identifying and validating drug targets and biomarkers by explicitly modeling uncertainty and probabilistic relationships.
Target Structure Confirmation
Confirming the precise 3D structure of a drug target is critical for rational drug design. Databases like the Potential Drug Target Database (PDTD), released in 2008, provide a valuable resource, containing over 1100 3D protein structures and covering 830 drug targets, complete with associated diseases, biological functions, and regulating pathways.
Traditionally, protein structural resolution relies on experimental methods such as Nuclear Magnetic Resonance (NMR) spectroscopy, X-ray crystallography, and cryo-electron microscopy (cryo-EM). While these techniques offer invaluable insights into protein structures and drug receptors, they are often expensive, time-consuming (ranging from weeks to years depending on sample availability and protein complexity), and require specialized equipment and expertise. AI, particularly tools like AlphaFold, has begun to complement these methods by providing highly accurate computational predictions of protein structures, significantly accelerating the initial stages of target characterization and reducing the reliance on purely experimental approaches for generating structural hypotheses.
Virtual Screening
Virtual screening (VS) is a computational technique used to rapidly search large libraries of chemical compounds for potential drug candidates.
Structure-based virtual screening (SBVS), also known as target-based VS, uses the 3D structure of a target protein to predict interactions with a library of compounds. Compounds are ranked based on their predicted affinity for the target’s receptor binding site. Molecular docking, a core SBVS technique, examines the geometric and energetic compatibility between ligands (drug candidates) and targets. Despite its widespread use, the complexity of ligand-receptor binding interactions can lead to difficulties in accurately predicting binding sites and classifying compounds, resulting in high false-positive and false-negative rates. The accuracy of SBVS heavily depends on the underlying search algorithms and scoring functions.

Ligand-based virtual screening (LBVS), conversely, identifies bioactive compounds by analyzing the chemical and structural features of known ligands, without requiring the target’s 3D structure. Traditional LBVS methods include molecular similarity searches and QSAR modeling. Recent advances in AI, particularly ML and DL techniques, have profoundly enhanced LBVS by improving molecular representation, optimizing similarity search algorithms, and refining the accuracy of QSAR models. AI-powered frameworks have also addressed challenges like data scarcity by incorporating multi-task learning, transfer learning, and graph-based neural networks, enabling more robust predictions even with limited experimental data.
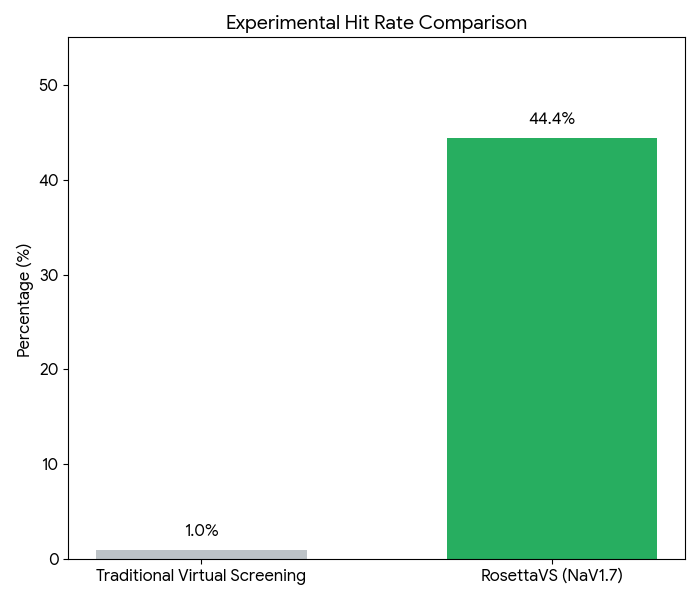
AI-based VS models often utilize molecular descriptors from physicochemical properties or fingerprints to build regression or classification models of activity, offering greater flexibility than traditional LBVS. Deep learning-based molecular docking, for instance, employs advanced AI algorithms to accelerate docking simulations and significantly improve the accuracy of pose predictions. Platforms such as BioNeMo, DiffDock, and RosettaVS leverage DL in combination with other computational methods to screen vast numbers of molecules with unprecedented speed and precision. A compelling case study using RosettaVS on the NaV1.7 target involved triaging a multi-billion compound space with active learning, docking 4.5 million compounds, synthesizing 9 candidates, and observing 4 micromolar binders—a remarkable 44.4% hit rate among synthesized compounds, demonstrating the power of these integrated AI workflows.
Synthetic Routes
Once a promising drug candidate is identified, determining the most efficient and cost-effective synthetic route for its production is a critical next step. Computer-Aided Synthetic Planning (CASP) tools assist chemists in this complex task, predicting selectivity, identifying potential byproducts, and suggesting optimal reaction conditions. CASP has evolved significantly, moving from systems based on manually coded reaction rules and templates to sophisticated AI-assisted synthesis planning. Modern AI algorithms can recommend synthetic routes for a wide range of reactions, even without explicit reaction templates, by learning from vast datasets of chemical reactions. These algorithms utilize various molecular representations, including fingerprints, graphs, or SMILES strings, to ‘understand’ chemical transformations.
Early CASP systems primarily used rule-based approaches, relying on coded rules and heuristics extracted from reaction databases and scientific literature, often referred to as "template methods." However, this approach is inherently limited by its inability to scale with the exponential growth of chemical literature and the incomplete nature of manually curated knowledge bases. Inverse synthesis software like Synthia addresses these limitations by combining powerful computational methods with an extensive, expert-coded rule base accumulated over 21 years (now over 115,000 rules) and a catalog of more than 12 million commercially available starting materials. Synthia employs a scoring function and a dynamic planning algorithm to construct synthetic pathways, making informed decisions at each inverse synthetic step and generating comprehensive proposals of synthetic routes for target molecules.
Complementing template-based methods, a template-free approach has emerged, drawing inspiration from NLP. This method frames synthetic prediction as a sequence-to-sequence (seq-2-seq) mapping problem, representing chemical reactions as "sentences" using SMILES strings of molecules. This treats the process as a form of chemical language translation, allowing AI models to "learn" the grammar and vocabulary of chemical reactions and predict novel synthetic pathways without explicit rules.
Revolutionizing Clinical Trials
Clinical trials represent the most expensive and time-consuming phase of drug development, with failure rates reaching as high as 95% in certain disease areas, particularly oncology. The majority of the $2.3 billion total cost of developing a new entity, approximately $1.4 billion, is attributed to in vivo studies. This highlights that while computational improvements in early drug discovery are crucial, their impact on overall cost reduction can be minimal if late-stage failures persist.
Therefore, models capable of predicting late-stage clinical trial outcomes could have a profound impact. Concepts such as the Virtual Physiological Human (VPH), first proposed in 2005, envision using virtual twin technology to support clinical decision-making and predict safety and efficacy. However, accurately modeling the intricate biology of the human organism remains a formidable challenge, and the accuracy of in silico clinical trial predictions requires further rigorous study.
In the interim, AI technologies are primarily focused on integrating diverse datasets—patient genetic data, electronic health records (EHRs), medical literature, and clinical trial databases—to predict trial success. Other AI models are designed to optimize trial design, enhance patient matching and recruitment, and improve patient adherence during trials.
Toxicity is a frequent cause of clinical trial failures, and computational models are proving effective in predicting adverse events. PrOCTOR, a tree-based ensemble ML model, combines chemical features of drugs with target-based features to distinguish between approved drugs and those that failed due to toxicity. Drugs predicted as toxic by PrOCTOR consistently showed significantly more frequent reports of serious adverse events than those predicted as safe. Similarly, Insilico Medicine’s inClinico platform utilizes deep neural networks based on pathway analysis to predict drug side effects by analyzing transcriptional changes induced by drugs. It forecasts clinical trial outcomes for 46 different side effects and provides a probability of success for a trial, identifying weak points in trial design for optimization. Other companies like Unlearn.ai, Saama, and Phesi offer similar predictive services, aiming to de-risk clinical development.
Natural Language Processing (NLP) techniques have also been instrumental in extracting crucial information from Electronic Medical Records (EMRs) to match patients to suitable clinical trials. NLP-based trial matching systems, notably demonstrated by IBM Watson teams, create detailed clinical profiles for patients and compare them against the complex eligibility requirements of available trials, significantly accelerating recruitment and ensuring a better fit.
Beyond prediction and recruitment, AI is revolutionizing patient adherence monitoring. Historically, trials relied on imprecise methods like pill counts and self-reported data. Now, AI systems are improving accuracy. AbbVie, for example, implemented an AI-powered facial and image recognition system through the AiCure mobile program, requiring patients to upload videos of themselves swallowing pills. The AI system verifies that the correct person took the prescribed medication. A study involving schizophrenia patients, a population often challenged with adherence, found that patients monitored with AI achieved an adherence rate of 89.7% over 24 weeks, significantly higher than the 71.9% observed in those monitored with modified directly observed therapy (mDOT), and a dramatic improvement over the typical 50% adherence rate for schizophrenia patients.
The Road Ahead: Challenges and Future Outlook
Digital and simulation tools have been steadily improving the drug discovery process since the 1980s, evolving from early CADD to the comprehensive data provided by the PDB, and culminating in the deep integration of AI in the 2020s. These advancements have undeniably contributed to bringing new, more effective medicines to market faster and more efficiently.
However, the rapid emergence of AI also presents significant challenges that must be addressed for its full potential to be realized in such a high-stakes industry. Trust in generative AI models remains a critical issue, largely due to the problem of "hallucinations" and the potential for misinformation. AI models are often regarded as "black boxes," providing accurate predictions without transparently revealing their decision-making processes. In healthcare, where patient safety and efficacy are paramount, transparency and explainability are as crucial as accuracy.
Furthermore, AI models typically require vast, high-quality datasets for effective training. This poses a considerable challenge, especially in rare disease areas where data is inherently scarce. For image recognition and NLP systems, achieving high performance often demands millions or even billions of data samples, which must be meticulously cleaned and curated—a time-consuming and resource-intensive endeavor. Even with seemingly clean data, inherent biases can inadvertently propagate into models from training data, leading to skewed predictions or recommendations that exacerbate existing disparities based on race, gender, or other demographic factors. Addressing these biases through careful data selection and algorithmic fairness is essential to ensure equitable healthcare outcomes.
The environmental footprint of AI is another growing concern. The training and operation of large AI models consume vast amounts of electricity and water, contributing significantly to carbon emissions. As the pharmaceutical and healthcare industry already faces pressures to reduce its environmental impact, the continued integration of AI necessitates a focus on sustainable AI practices and energy-efficient computational infrastructure.

Despite these formidable challenges, the promise of AI in significantly decreasing the timelines and costs associated with drug discovery and development is undeniable. By accelerating every stage from target identification to clinical trials, AI holds the potential to bring life-changing drugs to patients faster and more affordably. While ongoing research and ethical considerations are paramount, there is no doubt that these revolutionary methods will fundamentally transform the field of drug discovery and development, ultimately benefiting global health.









Leave a Reply