
The pharmaceutical industry faces an unprecedented challenge: the relentless rise in drug development costs and timelines, often referred to as "Eroom’s Law" (Moore’s Law spelled backward). Traditionally, bringing a new drug from initial discovery to market approval can take between 10 to 14 years, and sometimes even longer. This protracted process is coupled with astronomical financial investment, ranging from hundreds of millions to multiple billions of dollars per successful drug. Alarmingly, the inflation-adjusted cost of drug development has roughly doubled every nine years, exacerbating the pressure on pharmaceutical companies and healthcare systems alike.
The Mounting Costs and High Stakes of Drug Development
The exact research and development (R&D) cost per approved drug is notoriously difficult to pinpoint, varying significantly based on methodology, what elements are included (such as the cost of capital, failures, and post-approval work), and whether confidential firm data or public datasets are used. For instance, one influential analysis leveraging confidential company data estimated costs at approximately $1.4 billion out-of-pocket and $2.6 billion capitalized (in 2013 dollars). In contrast, a public-data analysis of FDA approvals placed the median capitalized R&D investment for a new drug at $985.3 million and the mean at $1335.9 million in its base case. Regardless of the precise figure, the consensus is that drug development is an exceedingly expensive endeavor.
Compounding the financial burden is the staggeringly low success rate. Only a small fraction of compounds entering preclinical testing ever progress to first-in-human studies. Furthermore, a mere 12% of drugs that advance to clinical trials ultimately gain FDA approval. The financial ramifications of late-stage trial failures are immense, with sunk development costs for a single failed trial ranging from $800 million to $1.4 billion.

Clinical development success rates are not uniform; they fluctuate by disease area and data source. A comprehensive benchmark of programs from 2011 to 2020 indicated an overall likelihood of approval from Phase I to market of approximately 7.9%. Oncology, a particularly challenging field, saw even lower rates, around 5.3%. Other industry summaries place the overall figure under 14%, underscoring the formidable hurdles faced by potential therapies.
This economically driven landscape has inadvertently created a profound gap in medical needs, especially for patients suffering from rare diseases. While 3.5% to 5.9% of the world’s population – an estimated 263 to 446 million people – are affected by rare diseases, a staggering 95% of them lack FDA-approved treatment options in the U.S. The high failure rates and associated financial losses compel pharmaceutical companies to prioritize more common diseases that promise larger market returns, leaving millions with rare and complex conditions without viable therapeutic avenues. This creates a vicious cycle: the prohibitive cost of failure concentrates investment on "blockbuster" indications, neglecting vast populations with unmet medical needs.
The Emergence of Digital Drug Design and AI: A Paradigm Shift
In response to these escalating challenges, digital drug design tools, powered increasingly by artificial intelligence (AI) and machine learning (ML), are emerging as a transformative force. These technologies are beginning to disrupt the traditional drug discovery paradigm by compressing timelines, substantially reducing costs, and expanding the range of diseases that are economically feasible to pursue. This shift promises to accelerate the delivery of life-saving medications and address the critical unmet needs of patients worldwide.
A Historical Journey: The Evolution of Digital Drug Design

To fully appreciate the current acceleration, it’s crucial to trace the lineage of digital drug design. Before the 1970s, drug discovery was largely a game of chance, dominated by serendipitous observations and extensive trial-and-error screening. Scientists, with a nascent understanding of molecular mechanisms, relied heavily on phenotypic screening and empirical testing. This often took decades and consumed substantial resources with no guarantee of success, making the process highly inefficient and unpredictable.
The 1970s marked a pivotal moment with the conceptualization and gradual emergence of computer-aided drug design (CADD). This revolutionary approach began integrating computational techniques to analyze the structure and interactions of molecules with drug targets. Early CADD applications, which gained traction throughout the 1980s, aimed to assess the activity, toxicity, and bioavailability of potential drug candidates in silico before costly physical experiments. This represented a significant leap from purely empirical methods towards a more rational and predictive approach.
The ensuing decades witnessed a rapid evolution and increasing accuracy in computational predictions. The 1980s saw the development of structure-based drug design (SBDD), which leverages the 3D structure of a target protein, and ligand-based drug design (LBDD), which focuses on the properties of known active compounds. The 2000s ushered in an era where structural biology, genomics, and bioinformatics became integral drivers of CADD development. The exponential growth of publicly accessible databases like the Protein Data Bank (PDB) and various chemical libraries provided vast datasets, making virtual screening a standard practice in early drug discovery.
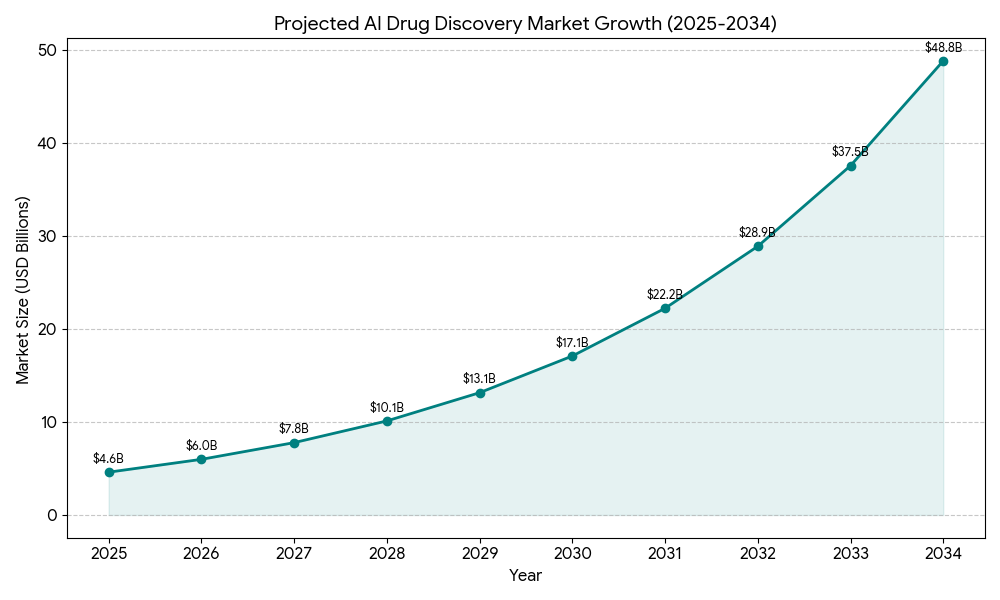
The 2020s represent the true inflection point, with the widespread integration of AI and ML into CADD. Deep learning models, capable of processing complex patterns in vast datasets, have enabled unprecedented predictive accuracy. Groundbreaking tools like AlphaFold, developed by DeepMind, have revolutionized protein structure prediction, and other generative AI tools are transforming molecular design by proposing entirely novel molecular structures. This rapid advancement is reflected in market projections: the AI drug discovery market is estimated to grow from $4.6 billion in 2025 to a staggering $49.5 billion by 2034, at a compound annual growth rate (CAGR) of 30%. This meteoric rise underscores the industry’s profound confidence in AI’s transformative potential.
The Present Moment: AI’s Broad Impact Across the Pipeline
Today, "digital drug design" is an expansive and rapidly evolving field. It encompasses a diverse toolkit, including sophisticated molecular simulations that model how compounds interact with biological targets at an atomic level, advanced machine learning models trained on vast chemical libraries to predict the properties and activity of drug candidates, and generative AI that can propose entirely novel molecular structures from scratch. Increasingly, these computational predictions are seamlessly integrated with lab automation platforms, creating a closed-loop system that accelerates experimental validation.
Adoption of these digital tools has accelerated considerably in recent years, driven by both the imperative to reduce R&D costs and the demonstrated success of early AI-powered initiatives. The first AI-designed drugs are now progressing through clinical trials, a testament to the technology’s maturity and potential. Companies like Insilico Medicine, known for its end-to-end AI-driven drug discovery platform, Recursion, which combines AI with experimental biology, and Absci, leveraging AI for de novo protein design, are at the forefront, showcasing comprehensive AI pipelines from target identification to candidate nomination.
The rising R&D costs are not just a challenge but also a powerful motivator, forcing pharmaceutical companies to seek efficiency improvements. Successes in related fields, such as the development of GLP-1 agonists for diabetes and weight loss, demonstrate the significant return on investment (ROI) potential that digital tool adoption can unlock. Furthermore, platforms integrating multiple digital tools are continually emerging. AI-powered lab notebooks, such as Sapio’s ELaiN, are moving beyond mere record-keeping to facilitate active collaboration and intelligent data analysis, further streamlining the research process.
AI’s Transformative Role in Specific Drug Discovery Stages:
The impact of AI and digital tools is not limited to a single phase but permeates the entire drug discovery and development pipeline, fundamentally altering how scientists approach each stage.

1. Molecular Dynamics Simulations:
Molecular dynamics (MD) simulations have evolved significantly, moving beyond static structures to model the dynamic behavior of molecules and proteins over time. Deep learning (DL) models are central to this advancement. Convolutional neural networks (CNNs) are adept at processing image-like data from molecular structure diagrams to predict properties and activity. Recurrent neural networks (RNNs) can leverage sequence data to capture long-term dependencies within molecular sequences, thereby improving the accuracy of property predictions. Graph neural networks (GNNs), which can directly process molecular graphs, are particularly powerful for modeling the complex relationships between atoms and molecules and predicting their properties.
The Simplified Molecular Input Line Entry System (SMILES) is widely used for drug characterization due to its linear molecular representation, allowing SMILES strings to be processed directly as text. This makes it invaluable in DL models for tasks like inverse synthesis prediction using sequence-to-sequence (seq-2-seq) methods. Its ability to generate multiple representations of the same molecule also provides an advantage for data augmentation. Similarly, molecular fingerprints (MFPs), bit strings encoding structural or pharmacological properties, are frequently used as input features in DL-based drug-target interaction (DTI) prediction models, enabling more accurate similarity searches and quantitative structure-activity relationship (QSAR) analyses.
2. Drug Target Discovery:
Identifying the right drug target is the crucial first step. AI algorithms, when combined with systems biology approaches, can meticulously analyze vast multi-omics data (genomics, proteomics, metabolomics) and correlate it with patient clinical health information. This allows for the discovery of previously unidentifiable correlations and potential therapeutic targets. Natural language processing (NLP) methods further enhance this process by retrieving and analyzing unstructured data from scientific literature, patents, and clinical reports, uncovering potentially relevant biological pathways and disease mechanisms that might otherwise remain hidden.
The inherent complexity of biological systems often poses challenges in constructing stable models for disease classification or identifying reliable biomarkers. However, advanced techniques like Bayesian AI analysis are proving instrumental. By integrating molecular profiles and multi-omics data with clinical health information, these models can construct causal inference networks, offering a more robust approach to identifying novel targets and biomarkers.
3. Target Structure Confirmation:
Confirming the precise 3D structure of a drug target is paramount for rational drug design. While traditional experimental methods like nuclear magnetic resonance (NMR), X-ray crystallography, and cryo-electron microscopy (Cryo-EM) provide invaluable insights, they are often time-consuming, expensive, and dependent on sample availability and protein complexity, with timelines ranging from weeks to years.
Digital tools have significantly augmented this process. Databases like the Potential Drug Target Database (PDTD), released in 2008, provide over 1100 3D protein structures covering 830 drug targets, complete with related diseases, biological functions, and associated pathways. More recently, AI models like AlphaFold have revolutionized protein structure prediction, enabling scientists to predict highly accurate 3D structures from amino acid sequences in a fraction of the time and cost required by experimental methods. This computational leap accelerates the initial stages of SBDD, making the confirmation of target structures more efficient.
4. Virtual Screening:
Virtual screening (VS) is a computational technique used to rapidly search large libraries of chemical compounds for those that are most likely to bind to a drug target, thereby reducing the number of compounds that need to be experimentally tested.
- Structure-Based Virtual Screening (SBVS): Also known as target-based VS, this CADD method predicts interactions between a target protein and a library of compounds using the target’s 3D structure. It ranks compounds based on their predicted affinity for the target’s receptor binding site. Molecular docking, a key SBVS technique, examines the geometric compatibility between ligands and targets. While widely used, the complexity of ligand-receptor binding interactions can lead to difficulties in predicting binding sites accurately and classifying compounds, resulting in high false-positive and false-negative rates. The accuracy of SBVS heavily relies on sophisticated search algorithms and scoring functions.
- Ligand-Based Virtual Screening (LBVS): This approach identifies bioactive compounds by analyzing the chemical and structural features of known active ligands, without requiring the target’s 3D structure. Traditional LBVS methods include molecular similarity searches and QSAR modeling.
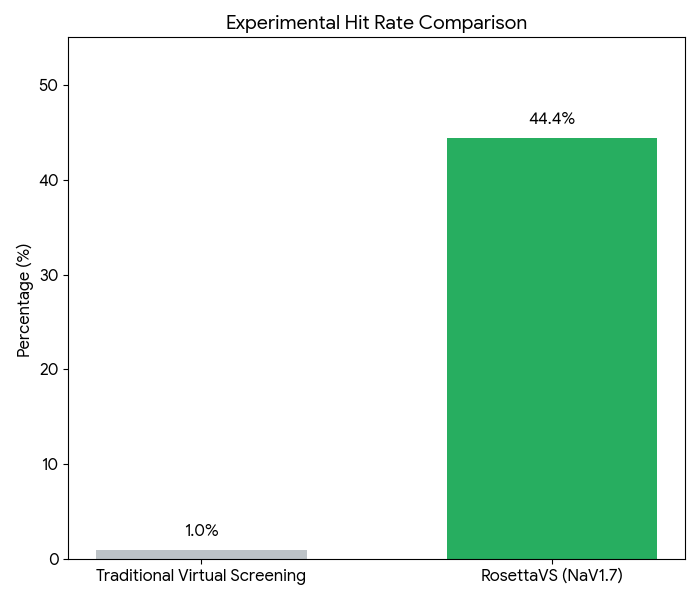
Advances in AI have dramatically enhanced both SBVS and LBVS. Machine learning and deep learning techniques have improved molecular representation, optimized similarity searches, and refined the accuracy of QSAR models. AI-powered frameworks help address data scarcity issues by incorporating multi-task learning, transfer learning, and graph-based neural networks. DL-based molecular docking, for instance, uses AI algorithms to accelerate docking simulations and improve the accuracy of pose predictions. Platforms like BioNeMo, DiffDock, and RosettaVS integrate deep learning with other methods to efficiently screen vast molecular libraries. A RosettaVS case study on NaV1.7 demonstrated the power of this approach: by triaging a multi-billion compound space using active learning, docking 4.5 million compounds, and synthesizing 9 candidates, the team achieved an impressive 44.4% hit rate (4 micromolar binders among synthesized compounds).
5. Synthetic Route Planning:
Identifying an efficient and cost-effective synthetic route is a critical, often bottlenecked, part of drug development. Computer-aided synthetic planning (CASP) tools assist chemists in this complex task by identifying optimal synthesis pathways, predicting selectivity and potential byproducts, and suggesting ideal reaction conditions. CASP has evolved significantly, moving from systems based on manually coded reaction rules and templates to sophisticated AI-assisted synthesis planning. AI algorithms can now recommend synthetic routes for a wide range of reactions, even those without pre-existing templates, by leveraging molecular representations like fingerprints, graphs, or SMILES strings.
Rule-based or "template methods" rely on coded rules and heuristics extracted from extensive reaction databases and literature. However, these methods are limited by their inability to scale with the exponential growth of chemical literature and the inherent incompleteness of human-curated knowledge bases. Inverse synthesis software like Synthia addresses these limitations by combining computational methods with expert-coded rules and a catalog of over 12 million commercially available starting materials. With a rule base accumulated over 21 years (exceeding 115,000 rules), Synthia uses a scoring function and dynamic planning algorithm to construct synthetic pathways, making informed decisions at each inverse synthetic step and generating proposals for multiple synthetic routes.
Complementing this, template-free approaches, drawing inspiration from NLP, frame synthetic prediction as a sequence-to-sequence mapping problem. By representing chemical reactions as "sentences" using SMILES strings, these models treat the process as a chemical language translation, allowing for the prediction of novel synthetic pathways without reliance on explicit rules.
6. Clinical Trials: A Major Frontier for AI
Clinical trials represent the most expensive and time-consuming stage of drug development, with failure rates from Phase 1 to approval in certain disease areas, such as oncology, reaching as high as 95%. Given that in vivo studies account for the majority of the cost of developing new entities (approximately $1.4 billion of a $2.3 billion total), computational improvements made in early drug discovery, while important, have only a minimal impact on overall cost reduction. This makes AI’s application in clinical trials potentially the most impactful for bringing down costs and accelerating drugs to patients.
Models that can accurately predict late-stage clinical trial outcomes could dramatically improve success rates. Concepts like the Virtual Physiological Human (VPH), proposed in a 2005 white paper, envision using virtual twin technology to support clinical decision-making and predict safety and efficacy. However, accurately modeling the complex biology of the human organism remains a significant challenge, and the accuracy of in silico clinical trial predictions requires further study.
Consequently, current AI technologies in clinical trials primarily focus on linking patient genetic data, electronic health records (EHRs), medical literature, and clinical trial databases to predict trial success. Other models are designed to improve trial design, assist with patient matching and recruitment, and monitor patient adherence.
- Toxicity Prediction: Toxicity is a frequent cause of clinical trial failures. Computational models can often predict this early. PrOCTOR, a tree-based ensemble ML model, combines chemical features of drugs with target-based features to distinguish between approved drugs and those that failed due to toxicity. Drugs predicted as toxic by PrOCTOR showed significantly more frequent reports of serious adverse events.
- Outcome Prediction Platforms: Insilico Medicine’s inClinico platform, for example, provides forecasts of a clinical trial’s probability of success. It can predict success rates, identify weak points in trial design, and optimize trials. Insilico has also developed a deep neural network (DNN) that predicts drug side effects by analyzing transcriptional changes induced by drugs, forecasting outcomes for 46 different side effects. Other companies like Unlearn.ai, Saama, and Phesi offer similar predictive and optimization services.
- Patient Recruitment and Adherence: NLP techniques are instrumental in extracting information from electronic medical records (EMRs) to create detailed clinical profiles, matching patients to the eligibility requirements of available clinical trials. This significantly speeds up recruitment, a common bottleneck. Furthermore, AI is revolutionizing patient adherence monitoring. Traditionally, trials relied on imprecise pill counts and self-reported data. Now, AI-powered systems, such as AbbVie’s implementation of AiCure’s facial and image recognition program, verify that the correct person takes the prescribed medication by requiring video uploads of pill ingestion. A study of schizophrenia patients demonstrated the profound impact: AI-monitored patients achieved an adherence rate of 89.7%, compared to 71.9% for those monitored with modified directly observed therapy (mDOT) over 24 weeks, a significant 17.9% difference. This is especially critical given that typical adherence rates for schizophrenia patients hover around 50%.
Addressing Unmet Medical Needs: A Renewed Hope

The economic shift brought about by AI in drug discovery has profound implications for neglected areas, particularly rare diseases. By drastically reducing the cost and time associated with early-stage discovery and increasing the likelihood of success, AI makes it economically viable to pursue drug candidates for diseases that were previously deemed too niche or risky. This offers a renewed hope for the millions of patients suffering from rare conditions who currently lack effective treatment options, potentially breaking the "vicious cycle" that has long plagued orphan drug development.
Challenges and Future Considerations
While the promise of AI in drug discovery is immense, significant challenges remain that must be addressed for its full potential to be realized.
- Trust and Transparency (The "Black Box" Problem): A central issue for generative AI models is low trust due to their inherent "black box" nature. While they may provide accurate predictions, understanding the rationale behind their decisions is often difficult. In a high-stakes industry like healthcare, where patient safety is paramount, transparency and explainability are crucial for regulatory approval and clinical adoption. Hallucinations and misinformation, although less common in scientific applications, remain a concern for overall trust.
- Data Requirements and Quality: AI models demand vast amounts of high-quality, clean, and diverse data for effective training. For image recognition and NLP systems, training often requires millions or even billions of data samples to achieve high performance. Sourcing, curating, and cleaning such datasets is a monumental task, often taking significant time and resources. In rare disease areas, where data is inherently scarce, this presents a particularly difficult hurdle.
- Bias in Data: Even with seemingly clean data, inherent biases can inadvertently creep into models from training datasets. Biases related to race, gender, socioeconomic status, or other demographic factors have been observed in various AI models. If left unaddressed, such biases in drug discovery AI could lead to drugs that are less effective or even harmful for certain patient populations, exacerbating existing health disparities.
- Environmental Impact: The operation of large AI models consumes a substantial amount of electricity and water. As the pharmaceutical and healthcare industry already faces scrutiny for its carbon footprint and waste generation, the continued, widespread integration of AI presents an environmental challenge that cannot be ignored. Sustainable AI practices and energy-efficient algorithms will be critical.
- Regulatory Frameworks: The rapid pace of AI innovation often outstrips the development of regulatory frameworks. Establishing clear guidelines for the validation, approval, and safe deployment of AI-designed or AI-optimized drugs and clinical trial processes is an ongoing challenge for regulatory bodies worldwide.
The Road Ahead
Despite these formidable challenges, the trajectory is clear: digital and simulation tools, dramatically enhanced by AI, are fundamentally transforming the drug discovery process. From the early days of CADD in the 1980s to the integration of the Protein Data Bank and the AI revolution of the 2020s, these advanced approaches have consistently helped bring new, more effective medicines to market faster and more efficiently. While the "black box" dilemma, data limitations, potential biases, and environmental concerns demand vigilant attention and ongoing innovation, there is little doubt that AI will continue to reshape the field, ultimately promising a future where life-changing treatments are discovered and delivered to patients with unprecedented speed and precision. The journey is complex, but the destination—a healthier, more equitable world—is well within sight.









Leave a Reply