
A groundbreaking study has identified a novel mechanism by which a single small molecule can leverage two distinct cellular pathways to degrade its target protein, offering a built-in backup system that promises to make targeted protein degraders significantly more effective and resistant to drug resistance. This discovery, detailed in research conducted by teams from the CeMM Research Center for Molecular Medicine, AITHYRA (both Institutes of the Austrian Academy of Sciences, Vienna, Austria), and the Centre for Targeted Protein Degradation (CeTPD; Dundee, UK), marks a pivotal advancement in the field of drug discovery, particularly for challenging diseases like cancer.
The Paradigm Shift: From Inhibition to Degradation
For decades, the vast majority of pharmaceutical drugs have operated on the principle of inhibition. These molecules typically bind to specific proteins, blocking their activity or function while leaving the protein structure itself intact. While highly successful in treating numerous conditions, this approach has inherent limitations. Many disease-driving proteins, especially in cancer, are considered "undruggable" by traditional inhibitors due to their lack of suitable binding pockets or their critical structural roles that cannot simply be inhibited without severe side effects. Moreover, cells can often adapt to inhibitory drugs by upregulating the target protein, activating alternative pathways, or developing mutations that render the inhibitor ineffective, leading to drug resistance.
Targeted Protein Degradation (TPD) represents a fundamental paradigm shift in pharmacology. Instead of merely blocking a protein’s activity, TPD molecules harness the cell’s own sophisticated quality-control machinery to completely remove the unwanted protein. This is achieved through bifunctional degrader molecules, often referred to as PROTACs (Proteolysis-Targeting Chimeras), which act as molecular matchmakers. One end of the degrader binds to the target protein, while the other end recruits an E3 ligase. E3 ligases are enzymes that label proteins with ubiquitin tags, marking them for destruction by the proteasome, the cell’s waste disposal system. By inducing this proximity, TPD drugs effectively hijack the cellular machinery to selectively eliminate disease-causing proteins.
The advantages of TPD are profound. It allows researchers to tackle previously "undruggable" targets, including scaffolding proteins or transcription factors whose functions are difficult to disrupt by inhibition alone. By completely eliminating the protein, TPD drugs can overcome resistance mechanisms that rely on protein overexpression and can disrupt both enzymatic activity and structural functions. The field has seen explosive growth over the past decade, with several TPD molecules now in clinical trials for various cancers and other diseases, demonstrating their therapeutic potential.
The Achilles’ Heel of Current Degraders: Single-Ligase Dependence
Despite its immense promise, a significant challenge in TPD has been the reliance of most degrader molecules on a single E3 ligase. The human genome encodes hundreds of different E3 ligases, each with specific roles in cellular protein homeostasis. When a TPD drug is designed to recruit only one particular E3 ligase, it creates a point of vulnerability. In the context of cancer, for instance, tumor cells are notoriously adaptable. They can acquire mutations in the targeted E3 ligase, downregulate its expression, or activate compensatory pathways to circumvent the drug’s mechanism, thereby developing resistance and rendering the treatment ineffective. This susceptibility to resistance has been a central hurdle in developing more durable TPD therapies.
A Molecular Backup System: The Dual-Ligase Discovery
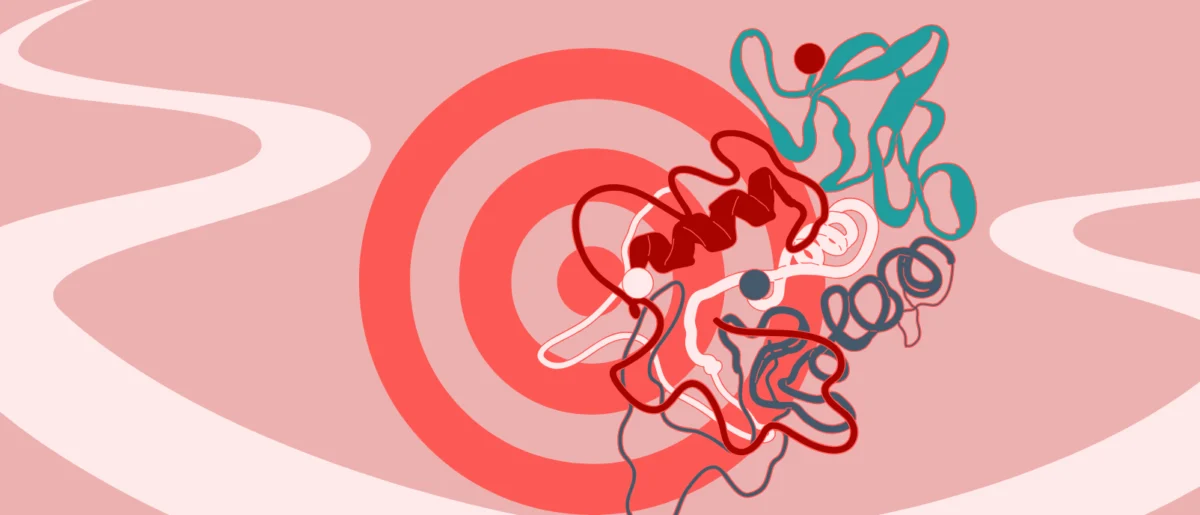
The new study, led by Georg Winter’s Group at CeMM and AITHYRA, in collaboration with Alessio Ciulli’s group at the CeTPD, addresses this critical limitation. The researchers investigated a small molecule designed to degrade SMARCA2/4, which are central ATPase subunits of the BAF chromatin remodeling complex. The BAF complex plays a crucial role in gene expression, and its dysregulation, particularly through mutations in SMARCA2/4, is frequently implicated in various cancers, making these proteins attractive therapeutic targets.
What the researchers uncovered was an unexpected and highly advantageous mechanism. Rather than engaging a single E3 ligase, the compound was found to simultaneously engage two distinct E3 ligase systems. Crucially, both pathways could independently drive the degradation of the target protein. The degradation process only came to a complete halt when both E3 ligase pathways were experimentally disabled. This revelation demonstrates a built-in molecular backup system, a feature rarely observed in synthetic drug mechanisms but common in natural biological systems, where redundancy serves to enhance robustness and resilience.
This dual engagement effectively provides a safety net. If one degradation pathway is compromised – for example, by a mutation in the E3 ligase or its reduced expression in a cancer cell – the other pathway can still ensure the efficient removal of the target protein. This redundancy significantly raises the bar for cancer cells to develop resistance, as they would need to adapt to or disable two distinct degradation mechanisms simultaneously, a much more challenging feat.
Unpacking the Mechanism: High-Definition Structural Insights

To understand how this dual-ligase engagement works at a molecular level, the research teams employed a sophisticated combination of genetic, biophysical, and structural deconvolution techniques. These methods allowed them to visualize and understand the intricate molecular interactions involved.
The findings revealed that the compound facilitates the formation of a highly specific ternary complex with its primary E3 ligase and the target protein. Simultaneously, the same molecule possesses the ability to recruit a second, distinct E3 ligase, providing an alternative route for the ubiquitination and subsequent degradation of SMARCA2/4. This dual-action capability highlights the intricate design possibilities for future degrader molecules.
A particularly striking finding was the "tunability" of this system. The researchers demonstrated that even subtle changes in the chemical structure of the compound could shift its preference from one E3 ligase to the other. Similarly, minor genetic alterations in the E3 ligases themselves could influence which pathway was preferentially engaged. This implies that ligase recruitment is not only dual but also highly adaptable and tunable, offering medicinal chemists unprecedented control over degradation pathways. This fine-tuning capability opens new avenues for optimizing drug efficacy and minimizing off-target effects.
Chronology of a Revolution: The Rise of Targeted Protein Degradation
The discovery of dual-ligase engagement represents an important evolutionary step in the relatively young field of targeted protein degradation. The concept of using small molecules to induce protein degradation first gained significant traction in the early 2000s with the pioneering work on PROTACs by research groups, including those led by Craig Crews and Raymond Deshaies. Initial proof-of-concept studies demonstrated that bifunctional molecules could indeed recruit E3 ligases to specific target proteins, leading to their degradation.
Over the past two decades, TPD research has accelerated dramatically. Key milestones include:
- Early 2000s: Initial development and characterization of PROTACs, demonstrating their ability to induce degradation.
- 2010s: Rapid advancements in linker chemistry and E3 ligase recruitment, leading to more potent and selective degraders. The discovery of cereblon-binding drugs like thalidomide and lenalidomide, which act as molecular glues to induce degradation, further expanded the TPD toolbox.
- Mid-2010s to Present: Expansion of E3 ligase targets beyond cereblon and VHL, increased understanding of TPD mechanisms, and the entry of numerous TPD candidates into preclinical and clinical development for various indications, particularly oncology. Several TPD drugs are now in advanced clinical trials, with some showing impressive preliminary results.
This latest discovery of a dual-ligase mechanism marks a significant leap forward, addressing one of the most pressing challenges in the field – overcoming resistance. It moves TPD from a primarily single-mechanism approach to one that incorporates biological redundancy and robustness, mirroring nature’s own strategies for resilience.
Expert Perspectives and Broader Implications
Georg Winter, co-corresponding author of the study, Life Science Director at AITHYRA, and Adjunct Principal Investigator at CeMM, emphasized the strategic importance of this discovery. "By enabling a single molecule to engage multiple degradation pathways, we can introduce redundancy into targeted protein degradation," Winter explained. "This could help overcome one of the key limitations of current degrader therapies, namely their susceptibility to resistance. It allows us to design drugs that are inherently more resilient."
Alessio Ciulli, co-corresponding author of the study and Director of the CeTPD, highlighted the profound impact of the structural insights gained. "This is an incredibly important development," shared Ciulli. "The structural detail we have been able to obtain here is remarkable. We can see precisely how this small molecule creates a new molecular handshake between proteins that would not normally interact. Because we can chemically tune which enzyme is doing the heavy lifting, medicinal chemists have a new avenue to explore when designing the next generation of cancer drugs. The collaboration between our two groups has once again proven to be a powerhouse for fundamental discovery."
The implications of this research extend far beyond the specific target protein SMARCA2/4. Drug resistance remains one of the most formidable obstacles in modern medicine, particularly in cancer therapy. According to the American Cancer Society, drug resistance is a major cause of treatment failure in approximately 90% of patients with metastatic cancer. This issue not only diminishes patient outcomes but also drives up healthcare costs as patients cycle through increasingly ineffective therapies. By distributing the degradative activity across multiple pathways, dual-ligase strategies could make it significantly more difficult for cancer cells to evolve resistance mechanisms, thereby prolonging the efficacy of treatments and improving patient survival rates.
This study fundamentally expands the conceptual framework of targeted protein degradation. It suggests that future drugs may not only be designed for exquisite specificity – binding precisely to their target – but also for inherent resilience and durability. This paradigm shift towards "durability by design" implies a new generation of medicines that are capable of maintaining their function and efficacy even as biological systems adapt and evolve, as is common in diseases like cancer.
The potential impact on pharmaceutical development is immense. Researchers can now explore designing degraders that deliberately recruit multiple E3 ligases, either to enhance potency, broaden the therapeutic window, or, critically, to preemptively counter resistance. This could lead to more robust therapies not only for cancer but also for a wide range of other diseases where protein dysregulation plays a role, such as neurodegenerative disorders and infectious diseases. Further research will focus on translating these findings into clinical applications, exploring the full spectrum of E3 ligase combinations, and optimizing the chemical properties of dual-ligase degraders for maximum therapeutic benefit. This discovery heralds a new era for targeted protein degradation, promising more powerful and enduring treatment options for patients worldwide.









Leave a Reply