
In a significant advancement for neurobiology, researchers have established a direct cause-and-effect link between mitochondrial dysfunction and the cognitive symptoms associated with neurodegenerative diseases, such as Alzheimer’s and other forms of dementia. A collaborative study published in Nature Neuroscience by teams from Inserm, the University of Bordeaux at the NeuroCentre Magendie, and the Université de Moncton in Canada suggests that the failure of cellular "power plants" is not merely a byproduct of brain aging or disease, but a primary driver of the memory loss and cognitive decline that characterize these conditions. By utilizing a sophisticated new molecular tool to artificially boost mitochondrial activity in animal models, the research team demonstrated that restoring energy production in neurons can significantly improve memory performance, potentially shifting the paradigm for how future dementia treatments are developed.
The Energetic Demands of the Human Brain
To understand the implications of this study, one must first consider the unique metabolic profile of the human brain. While the brain accounts for only approximately 2% of the average adult’s body weight, it consumes nearly 20% of the body’s total oxygen and glucose. This disproportionate energy requirement is driven by the constant need for neurons to maintain ion gradients, facilitate synaptic transmission, and repair cellular structures. Within these neurons, mitochondria serve as the primary site of adenosine triphosphate (ATP) production through oxidative phosphorylation.
In a healthy brain, mitochondria are dynamic organelles, constantly moving to areas of high energy demand, such as synapses, where they fuel the release of neurotransmitters. When these organelles fail, the resulting "energy crisis" leaves neurons unable to communicate effectively. The recent findings suggest that this bioenergetic failure occurs early in the progression of neurodegenerative diseases, often preceding the widespread death of brain cells. This insight provides a critical window for intervention, suggesting that if the energy supply can be stabilized, the functional decline of the brain might be slowed or even reversed before irreversible damage occurs.
A Chronology of Mitochondrial Research in Neurodegeneration
The path to this discovery has been paved by decades of observation and shifting scientific hypotheses regarding the origins of dementia.
- Early Observations (1906–1980s): Since Alois Alzheimer first described the plaques and tangles associated with the disease that bears his name, research primarily focused on protein misfolding. However, as early as the mid-20th century, pathologists noted that the brains of deceased dementia patients often showed structural abnormalities in mitochondria, though these were largely dismissed as late-stage "cellular debris."
- The Amyloid Cascade Hypothesis (1990s–2010s): For nearly thirty years, the dominant theory suggested that the accumulation of amyloid-beta plaques was the primary cause of Alzheimer’s. While this led to numerous clinical trials, many failed to show significant cognitive improvement, leading researchers to look for earlier triggers.
- The Bioenergetic Turn (2010–2020): Evidence began to mount that metabolic changes, such as reduced glucose metabolism (often called "Type 3 Diabetes"), were visible in PET scans of patients years before the onset of clinical symptoms. Studies linked mitochondrial "Complex I" disruptions to the very earliest stages of cognitive impairment.
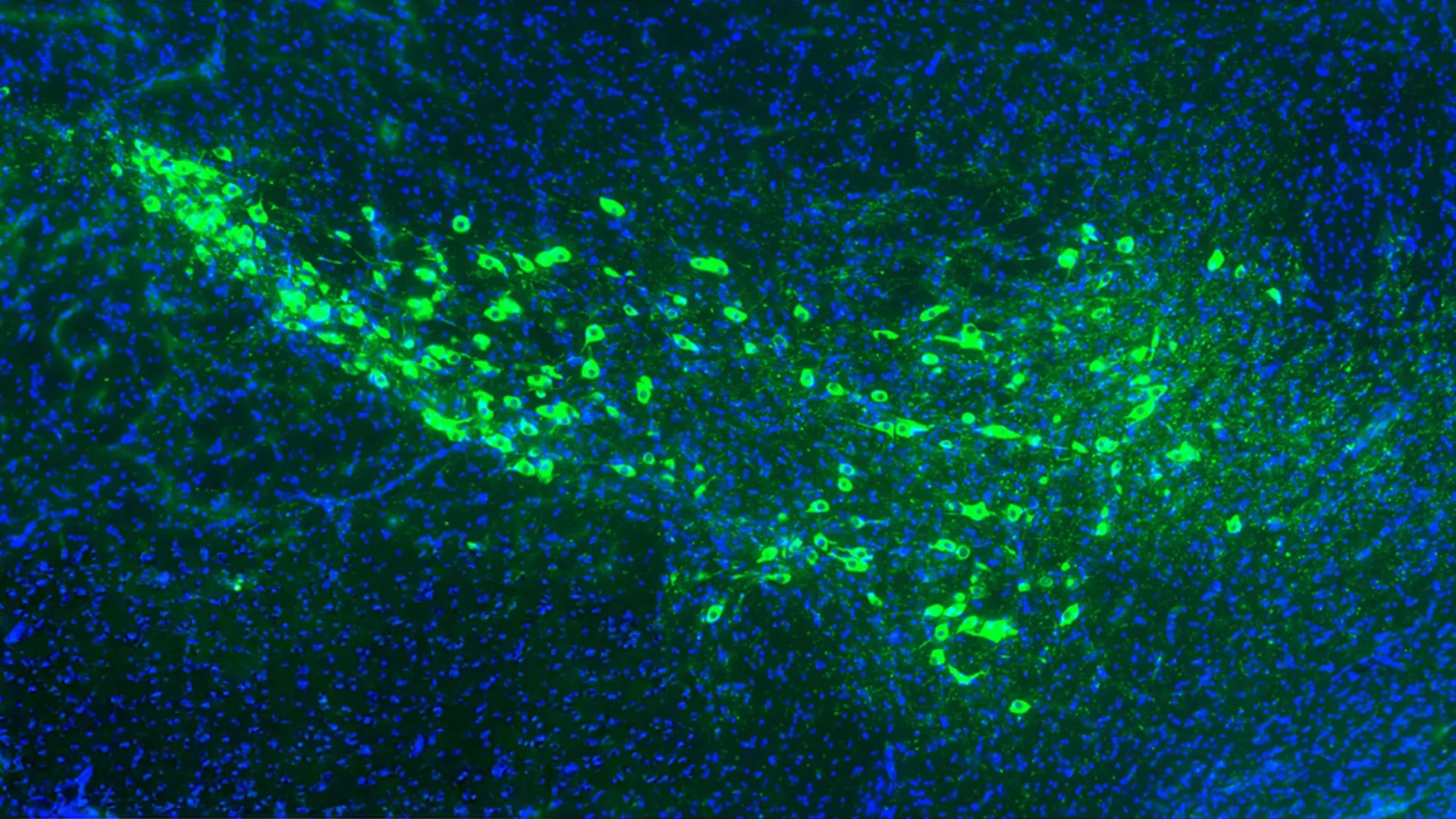
- The 2025 Breakthrough: The current study by Marsicano, Hébert Chatelain, and Bellocchio moves beyond observation to intervention. By creating the "mitoDreadd-Gs" tool, researchers moved from asking "are mitochondria broken?" to "can fixing them fix the memory?"
Technical Innovation: The Development of mitoDreadd-Gs
The centerpiece of this research is a highly specific chemogenetic tool known as mitoDreadd-Gs. Previously, scientists struggled to manipulate mitochondrial activity without affecting other parts of the cell or causing systemic toxicity. The research teams leveraged their knowledge of G proteins—signaling molecules that act as internal switches within cells—to create an artificial receptor.
This receptor, mitoDreadd-Gs, was engineered to be expressed specifically within the mitochondria of neurons. When activated by a specific, otherwise inert drug, the receptor stimulates the G proteins located on the mitochondrial membrane. This activation triggers a cascade that increases the organelle’s efficiency in producing ATP.
In the experiments conducted on mouse models of dementia, the researchers observed that when the mitoDreadd-Gs tool was "switched on," the mitochondrial activity in the hippocampus—the brain’s primary memory center—returned to levels seen in healthy control groups. More importantly, this physiological restoration was accompanied by a measurable recovery in cognitive function. The mice, which previously struggled with spatial memory and recognition tasks, showed a significant improvement in their ability to navigate and remember new information.
Supporting Data and Collaborative Findings
The Inserm and University of Bordeaux study does not stand in isolation. It aligns with recent data from the Mayo Clinic, which utilized proteomic and metabolomic profiling to link disruptions in mitochondrial Complex I—the first enzyme in the respiratory chain—to the progression of Alzheimer’s. The Mayo Clinic’s research highlighted that patients with more robust mitochondrial function tended to show a slower rate of cognitive decline, even in the presence of amyloid plaques.
Furthermore, reviews of longitudinal aging studies suggest that mitochondrial DNA mutations and oxidative stress are among the most reliable biomarkers for predicting the transition from mild cognitive impairment to full-scale dementia. The data suggests a "threshold effect": the brain can compensate for a certain level of protein buildup, but once mitochondrial output falls below a specific percentage of the required energy, the synaptic network begins to collapse.
Perspectives from the Research Leaders
The researchers involved in the study emphasize that while the results are promising, the transition from animal models to human therapies remains a complex challenge.
"This work is the first to establish a cause-and-effect link between mitochondrial dysfunction and symptoms related to neurodegenerative diseases, suggesting that impaired mitochondrial activity could be at the origin of the onset of neuronal degeneration," stated Giovanni Marsicano, Inserm research director and co-senior author. His statement underscores the shift from viewing mitochondria as victims of disease to viewing them as potential architects of the disease process.
Étienne Hébert Chatelain, a professor at the Université de Moncton and co-senior author, highlighted the diagnostic and therapeutic potential of the new tool. "Ultimately, the tool we developed could help us identify the molecular and cellular mechanisms responsible for dementia and facilitate the development of effective therapeutic targets," he explained.
Luigi Bellocchio, an Inserm researcher and co-senior author, noted that the next phase of research will focus on the long-term viability of this approach. "Our work now consists of trying to measure the effects of continuous stimulation of mitochondrial activity to see whether it impacts the symptoms of neurodegenerative diseases and, ultimately, delays neuronal loss or even prevents it," Bellocchio added.
Analysis of Implications for Future Therapeutics
The discovery that memory loss is tied to "living neurons running short on energy" rather than exclusively to "dead neurons" offers a more optimistic outlook for dementia research. If the cognitive symptoms of dementia are, at least in part, a functional deficit caused by energy starvation, it implies that those neurons are still salvageable.
This could lead to a new class of "bioenergetic" therapies. Current Alzheimer’s drugs, such as lecanemab, focus on clearing amyloid plaques from the brain. While effective at plaque removal, their impact on cognitive recovery has been modest. A dual-action approach—clearing toxic proteins while simultaneously "recharging" the neuronal energy supply—could prove far more effective.
Moreover, this research has implications beyond Alzheimer’s. Mitochondrial dysfunction is a hallmark of Parkinson’s disease, Huntington’s disease, and Amyotrophic Lateral Sclerosis (ALS). The mitoDreadd-Gs tool provides a blueprint for investigating whether boosting energy production could serve as a universal neuroprotective strategy across various forms of neurodegeneration.
Challenges and Ethical Considerations
Despite the excitement, several hurdles remain. Delivering genetic tools like mitoDreadd-Gs to the human brain requires gene therapy vectors, which are currently expensive and carry risks of immune reactions. Additionally, there is the question of "metabolic exhaustion." Scientists must determine whether forcing mitochondria to work harder over a long period might eventually lead to increased oxidative stress or "burnout" of the organelles themselves.
There is also the matter of timing. If mitochondrial failure is indeed an early event, then screening for metabolic health in the brain must become a priority. This would require more widespread use of advanced imaging techniques, such as functional MRI (fMRI) or specialized PET scans, to identify individuals at risk before they show outward signs of memory loss.
Conclusion: A New Frontier in Brain Health
The study from Inserm, the University of Bordeaux, and the Université de Moncton represents a pivotal moment in the fight against dementia. By proving that mitochondrial activity directly dictates cognitive health, the researchers have provided a tangible target for the next generation of medical interventions.
As the global population ages, the burden of neurodegenerative disease is expected to triple by 2050. The traditional focus on the "hallmarks" of the disease—the plaques and tangles—is now being supplemented by a deeper understanding of the cellular machinery that keeps the brain alive. By learning how to recharge the tiny engines within our cells, science may finally be moving toward a future where the "power failure" of dementia can be prevented, ensuring that the brain’s lights stay on for longer.









Leave a Reply