
Covalent drugs, a class of therapeutics that form irreversible or quasi-irreversible bonds with their biological targets, have undergone a remarkable resurgence in drug discovery and development. From the early, often serendipitous discoveries of agents like aspirin and penicillin to the sophisticated, rationally designed targeted covalent inhibitors (TCIs) of today, these molecules offer distinct advantages, particularly in achieving sustained pharmacological effects and addressing previously "undruggable" targets. However, their unique mechanism of action presents complex pharmacokinetic (PK) and pharmacodynamic (PD) considerations that necessitate a specialized approach throughout their development lifecycle. Understanding and effectively managing these distinctive properties is paramount for safely and efficiently bringing these transformative therapies to patients.
A Legacy of Transformation: The Historical Arc of Covalent Drugs
The journey of covalent drugs began long before their intricate mechanisms were fully deciphered. Aspirin, acetylsalicylic acid, approved in 1899, stands as a testament to early pharmacological success. Its anti-inflammatory, analgesic, and antipyretic properties, later attributed to the irreversible acetylation of cyclooxygenase (COX) enzymes, revolutionized pain management and cardiovascular disease prevention. Yet, it took nearly 70 years for Sir John Vane and his colleagues to elucidate aspirin’s precise covalent inhibition of COX-1 and COX-2, a discovery that earned him a Nobel Prize.
Similarly, penicillin, discovered serendipitously by Alexander Fleming in 1928 and subsequently developed into a life-saving antibiotic by Howard Florey and Ernst Chain in the early 1940s, marked a pivotal moment in medical history. Its ability to inhibit bacterial cell wall synthesis by forming a covalent bond with the serine residue of bacterial transpeptidase enzymes (penicillin-binding proteins) was not fully understood until more than five decades after its widespread clinical use. These early breakthroughs, while revolutionary, underscored a period where the "how" often preceded the "why" in drug development.
The Era of Caution: A Stalled Trajectory
Despite the undeniable success of aspirin and penicillin, the field of covalent drug development faced a significant slowdown in the latter half of the 20th century. The primary deterrent was a pervasive concern regarding the safety profile of electrophilic compounds, the chemical "warheads" typically employed to form covalent bonds with biological macromolecules. Early studies revealed that many electrophiles, particularly highly reactive ones, could bind indiscriminately to a wide array of proteins, lipids, and even DNA, leading to off-target toxicities, cellular damage, and, in some cases, carcinogenicity.
High-profile examples of toxicity associated with electrophilic metabolites reinforced this perception. Acetaminophen (paracetamol), for instance, can form a highly reactive electrophilic metabolite (N-acetyl-p-benzoquinone imine, NAPQI) that, when glutathione stores are depleted, binds covalently to hepatic proteins, causing severe liver damage. Other compounds like bromobenzene and the components of urushiol (the irritant in poison ivy) further illustrated the dangers of non-selective electrophilic reactivity. This led to a prevailing sentiment within medicinal chemistry that electrophiles represented "no-go" zones, pushing drug discovery predominantly towards reversible, non-covalent binding mechanisms, which were perceived as inherently safer due to their transient interactions and predictable dose-response relationships. Regulatory bodies also became increasingly stringent, demanding robust safety data and a clear understanding of drug mechanisms, which further complicated the development pathway for compounds with potentially promiscuous binding characteristics.
The Resurgence: Rational Design and Soft Electrophiles
The landscape began to shift around the turn of the 21st century, driven by advancements in structural biology, computational chemistry, and a deeper understanding of protein biochemistry. Researchers realized that not all electrophiles were created equal. The key lay in carefully tuning the electrophilicity of the "warhead" to achieve high selectivity and controlled reactivity. This led to the adoption of "soft electrophiles," such as acrylamides, nitriles, and sulfonyl fluorides, which react preferentially with specific, highly nucleophilic amino acid residues, particularly cysteine thiols, within defined binding pockets of target proteins.
This paradigm shift ushered in the era of rationally designed Targeted Covalent Inhibitors (TCIs). Instead of accidental discovery, modern covalent drug development employs sophisticated strategies to design compounds that form a covalent bond with a specific residue in the target protein, typically a cysteine, lysine, or histidine, while minimizing reactivity with other biological molecules. This targeted approach has dispelled the earlier belief that all electrophiles are inherently promiscuous and unsafe.
A striking example of this controlled reactivity is the comparison between sucralose, a safe artificial sweetener containing alkyl chlorides, and sulfur mustard, a chemical weapon that also contains alkyl chlorides. The difference lies in their precise chemical structure and the resulting electrophilicity, demonstrating how subtle modifications can drastically alter biological outcomes from benign to deadly.
The success of these lower-reactivity warheads has fueled a boom in covalent drug development. To date, over 110 covalent drugs have received regulatory approval across diverse therapeutic areas. Beyond the historical giants, modern examples include:
- Omeprazole: A proton pump inhibitor for gastrointestinal diseases, which covalently binds to cysteine residues on H+/K+-ATPase.
- Telaprevir: An HCV protease inhibitor, demonstrating efficacy against hepatitis C virus.
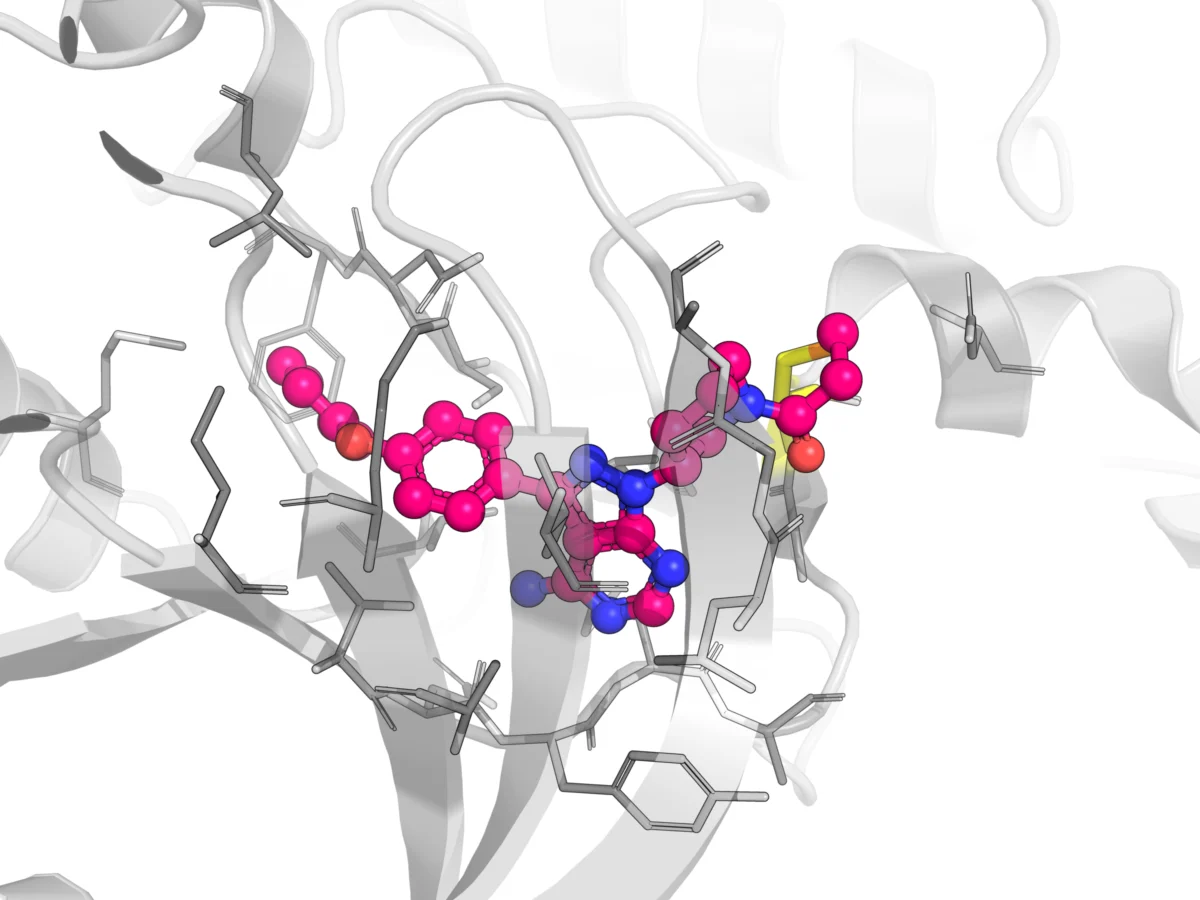
- Osimertinib (Tagrisso): A third-generation EGFR inhibitor, critically important for treating non-small cell lung cancer with specific EGFR mutations, particularly those resistant to earlier therapies. It covalently binds to Cys797 in the EGFR kinase domain.
- Ibrutinib (Imbruvica): An irreversible inhibitor of Bruton’s Tyrosine Kinase (BTK), revolutionizing the treatment of various B-cell malignancies like chronic lymphocytic leukemia and mantle cell lymphoma. It forms a covalent bond with Cys481 in BTK.
This resurgence has been accompanied by the development of specialized metrics for evaluating covalent drugs, including kinact/Ki (a measure of inactivation kinetics and affinity), assessment of glutathione (GSH) conjugation (indicating potential non-specific reactivity), and time-dependent inhibition (TDI) risk, all of which aim to characterize the drug’s reactivity and selectivity more comprehensively. Despite these advances, the unique covalent binding mechanism introduces distinct pharmacokinetic challenges that demand careful consideration.
Navigating the Unique Pharmacokinetic Landscape of Covalent Drugs
The irreversible or quasi-irreversible nature of covalent binding fundamentally alters the pharmacokinetic profile compared to conventional, reversibly binding small-molecule drugs. Key areas of divergence include protein binding beyond the target, novel clearance mechanisms, the propensity for time-dependent inhibition of cytochrome P450 enzymes, and a decoupled PK/PD relationship.
Protein Binding Beyond the Target Protein
While covalent binding to the intended biological target is crucial for therapeutic efficacy, covalent drugs can also engage in off-target binding with other proteins in the body. These non-specific interactions can significantly influence the drug’s distribution, metabolism, and potential for toxicity. For instance, covalent drugs may form stable adducts with abundant plasma proteins like human serum albumin (HSA). Such binding reduces the concentration of free drug in plasma, which is generally considered the pharmacologically active fraction. While high levels of HSA binding don’t automatically equate to adverse effects, they can impact distribution kinetics, alter the volume of distribution, and potentially contribute to clearance pathways by forming a larger, more stable complex that is processed differently by the body.
Beyond plasma, covalent drugs can also bind to tissue proteins, particularly in highly metabolic organs like the liver. This tissue binding can lead to drug accumulation, potentially influencing local metabolic processes, prolonging retention, and contributing to organ-specific toxicities if the off-target interactions are biologically active or lead to cellular dysfunction. The challenge for developers is to meticulously differentiate between on-target engagement, which is desired, and off-target reactivity, which can lead to toxicity. This requires careful optimization of the drug’s selectivity and reactivity profile during the lead optimization phase, often involving a balance between sufficient reactivity for target engagement and minimal reactivity for off-targets. Advanced proteomic techniques, such as activity-based protein profiling (ABPP), are increasingly employed to map the global reactivity profile of covalent inhibitors, providing crucial insights into their selectivity.
Conjugation and Protein-Adduct-Mediated Clearance Mechanisms
Covalent drugs often undergo distinct clearance pathways, primarily through conjugation reactions. Glutathione (GSH) conjugation and cysteine adduct formation represent major routes of metabolic inactivation for many electrophilic compounds. Glutathione, a ubiquitous tripeptide, acts as a primary cellular defense against electrophilic species, forming stable thioether conjugates that are subsequently processed and excreted. This pathway is often catalyzed by glutathione S-transferases (GSTs), a diverse family of enzymes with genetic polymorphisms that can influence an individual’s capacity for detoxification.
Consider Futibatinib, an irreversible FGFR1-4 inhibitor approved for FGFR2-rearranged cholangiocarcinoma. Its primary metabolism involves O-demethylation and significant glutathione conjugation. A mass-balance study revealed that a major circulating metabolite was a cysteinylglycine conjugate, representing approximately 13% of the circulating drug-related material. Further hepatocyte studies identified additional GSH, cysteine, glucuronide, and sulfate metabolites, underscoring the complexity of its metabolic fate.
Industry Best Practices for Clearance: Drug developers are advised to quantify both non-enzymatic and GST-mediated reactivity early in the discovery process. Ranking the electrophilic reactivity of compounds and utilizing recombinant GST isoforms and hepatocyte systems can help assess the potential risks associated with genetic polymorphisms in GST enzymes, which could impact drug clearance and efficacy in different patient populations. Furthermore, the elimination of covalent drugs can occur through the breakdown and clearance of drug-protein adducts, such as albumin-bound species. These adducts, once formed, can be subject to normal protein turnover processes, with the entire adduct being degraded and excreted. This "adduct-mediated clearance" can account for a substantial portion of total drug excretion and needs to be factored into PK modeling.
Time-Dependent Inhibition (TDI) of CYP450 Enzymes
A significant proportion of covalent drugs act as time-dependent inhibitors (TDIs) of at least one human cytochrome P450 (CYP) enzyme. CYP450 enzymes are crucial for the metabolism of numerous drugs, and their inhibition can lead to clinically significant drug-drug interactions (DDIs). Unlike reversible inhibition, TDI involves the formation of a stable, often irreversible, complex between the drug (or its metabolite) and the CYP enzyme, leading to a progressive loss of enzyme activity over time. This can result in an exaggerated or prolonged increase in the exposure of co-administered drugs that are substrates for the inhibited CYP enzyme.
The study by Moghaddam MF et al. (2014) highlighted this concern, evaluating ten approved covalent drugs across multiple CYP isoforms and finding that most exhibited time-dependent inhibition of at least one enzyme, indicating a pervasive TDI risk within this class. The mechanism often involves the drug’s electrophilic warhead, or a reactive metabolite thereof, covalently binding to the heme iron or the protein moiety of the CYP enzyme.
Industry Best Practices for TDI: Early kinetic characterization, including the determination of inactivation rate constant (kinact) and the concentration required to achieve half-maximal inactivation (KI), is critical. Mechanistic modeling of TDI can help predict the clinical significance of potential DDIs and guide structural optimization efforts to mitigate this risk. In cases where TDI cannot be eliminated, appropriate dosing strategies or warnings for co-administration with sensitive CYP substrates may be necessary during regulatory review.
The Decoupled PK/PD Relationship: Target Occupancy Reigns Supreme
Perhaps the most defining pharmacokinetic characteristic of covalent drugs is the decoupling of their plasma concentration (PK) from their pharmacological effect (PD). For traditional, reversible small-molecule drugs, continuous maintenance of drug concentrations above a certain threshold in the plasma (and at the target site) is essential to sustain the therapeutic effect. The duration of action is directly tied to the drug’s plasma half-life and its ability to remain bound to the target.
Covalent drugs, however, form a persistent bond with their target protein. Once this irreversible bond is established, the drug effectively "locks" the active site, and its therapeutic effect is sustained as long as the target protein remains inactivated. The recovery of target activity then becomes dependent not on the drug’s plasma half-life, but on the cellular synthesis rate of new, functional target protein. The drug molecule itself might be rapidly cleared from the plasma, but its effect persists.
A compelling example is the irreversible BTK inhibitor CC-292. Studies showed that plasma levels of CC-292 plummeted to near or below the lower limit of quantification within 24 hours post-dose. Yet, target occupancy of BTK in relevant cells remained high for up to 24 hours and only declined as new BTK protein was synthesized. Similarly, ibrutinib, despite a relatively short plasma half-life, achieves prolonged BTK inhibition due to its irreversible binding and the slow turnover of the BTK enzyme. This phenomenon profoundly impacts dosing strategies and the interpretation of PK data. Optimizing plasma exposure (e.g., maintaining high trough concentrations) becomes less critical than ensuring rapid and sufficient initial target engagement (Cmax-driven engagement) to achieve maximal target occupancy.
Industry Best Practices for PK/PD: To address this unique relationship, developers should shift their focus from traditional PK parameters (like AUC, Cmax, Tmax in isolation) to directly modeling target occupancy. This often involves the use of ex vivo or in vivo pharmacodynamic biomarkers that directly measure target engagement or downstream effects, such as the phosphorylation status of a protein or the inhibition of a cellular pathway. Dosing regimens should be designed to achieve rapid, saturating target binding, with the frequency of dosing determined by the target’s turnover rate rather than the drug’s systemic clearance. This necessitates a deep understanding of the biology of the target protein, including its synthesis and degradation rates in relevant tissues.
Broader Impact and Future Outlook
Covalent drugs represent a powerful class of therapeutics that address some of the most critical unmet medical needs, including overcoming drug resistance, targeting difficult-to-drug proteins, and achieving durable responses with potentially lower dosing frequencies. Their ability to provide prolonged target occupancy without continuous systemic exposure offers significant advantages in terms of efficacy and patient compliance.
The increasing sophistication in designing highly selective soft electrophiles, coupled with advanced analytical techniques for characterizing their binding kinetics and off-target reactivity, continues to accelerate development in this field. Beyond the current generation of TCIs, the principles of covalent chemistry are being extended to novel modalities like Proteolysis-Targeting Chimeras (PROTACs). PROTACs are bifunctional molecules that leverage covalent (or highly affine non-covalent) binding to bring a target protein into proximity with an E3 ubiquitin ligase, leading to the target’s ubiquitination and subsequent proteasomal degradation. This represents an even more advanced form of targeted protein engagement, offering new avenues for therapeutic intervention.
For developers, success in this rapidly evolving landscape hinges on a profound understanding of the unique pharmacokinetic properties of covalent drugs. Their development programs must demonstrate deliberate, selective chemistry, supported by a mechanistic understanding of both efficacy and safety. Regulatory bodies are increasingly sophisticated in their assessment of these compounds, requiring robust data on target engagement, off-target reactivity, and the decoupled PK/PD relationship. Organizations with specialized expertise in DMPK (Drug Metabolism and Pharmacokinetics), like WuXi AppTec where Dr. Qigan Cheng (a senior Study Director) contributes, are vital in guiding development teams through these intricate challenges, from early discovery to clinical translation. As technology continues to advance, the efficient and safe development of transformative covalent therapies will increasingly rely on this specialized knowledge and integrated approach. The future of covalent drugs promises to unlock even more therapeutic potential, solidifying their role as a cornerstone of modern medicine.







Leave a Reply