
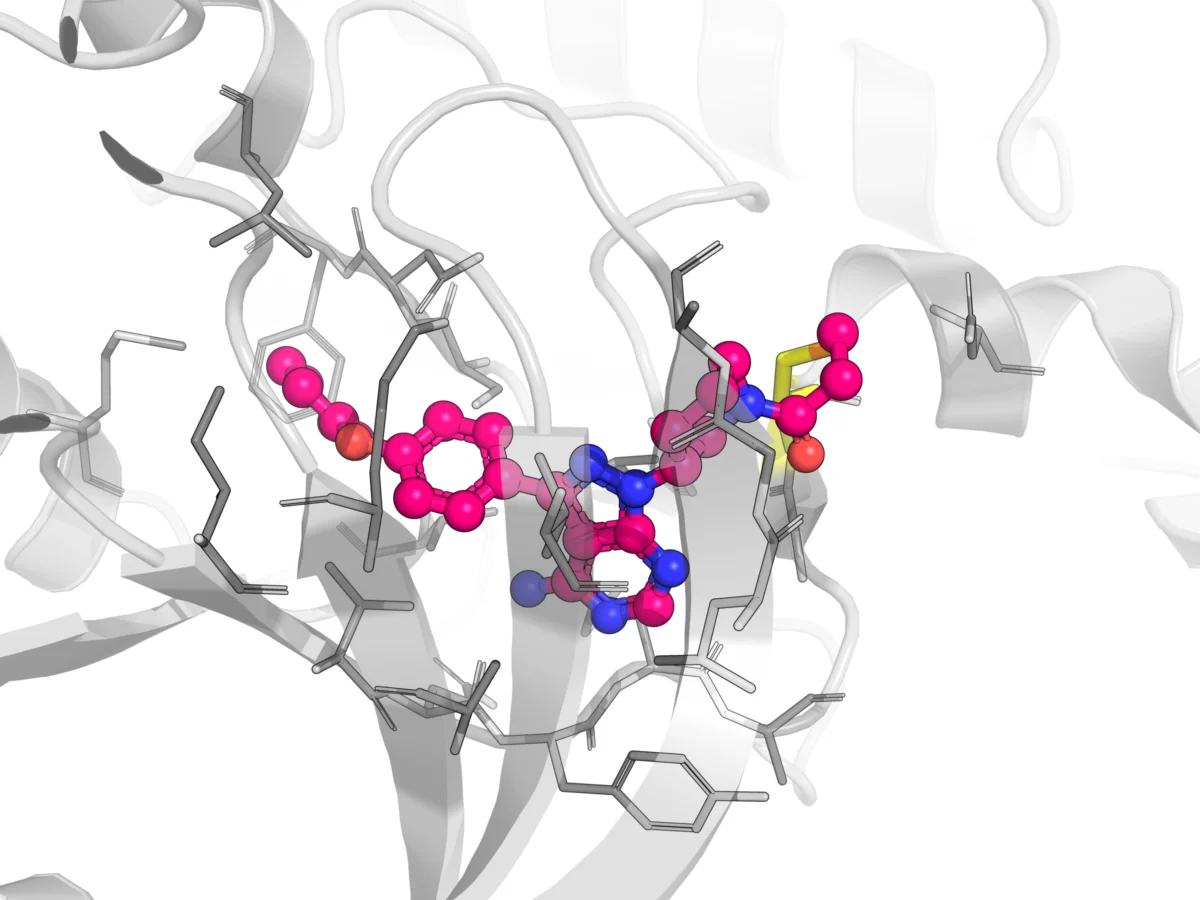
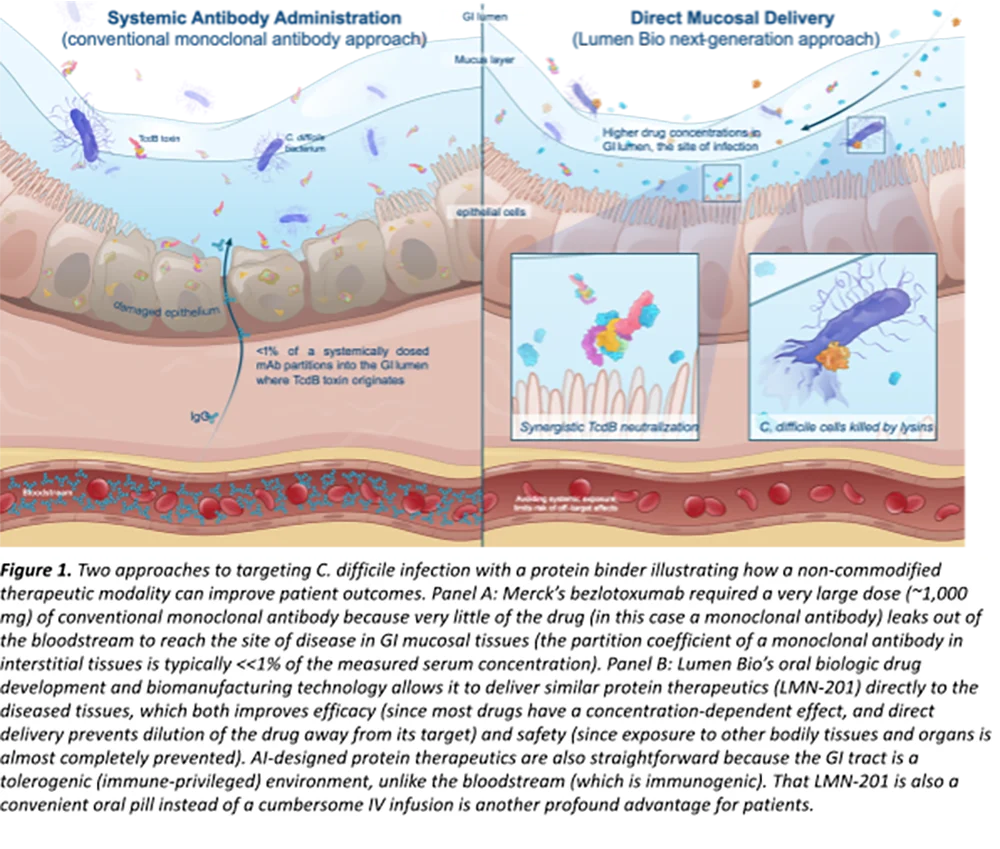
The landscape of modern therapeutics has been significantly shaped by covalent drugs, a class of pharmaceuticals distinguished by their ability to form stable, irreversible bonds with their biological targets. From the widespread adoption of aspirin in 1899 to the life-saving impact of penicillin, these agents have consistently pushed the boundaries of medical innovation. While early discoveries were largely serendipitous, a sophisticated understanding of molecular mechanisms and rational drug design has ushered in a new era for covalent inhibitors, particularly over the last decade. This resurgence, however, brings with it a unique set of pharmacokinetic (PK) challenges that demand a tailored approach to development, moving beyond traditional small-molecule drug evaluation paradigms.
A Legacy of Serendipity: The Early Days of Covalent Drug Discovery
The story of covalent drugs is deeply intertwined with some of the most profound medical breakthroughs in history. Aspirin, acetylsalicylic acid, was first synthesized by Felix Hoffmann at Bayer in 1897 and approved for medical use shortly thereafter. Its widespread analgesic, anti-inflammatory, and antipyretic properties made it a staple in medicine cabinets globally. Yet, its precise mechanism of action remained a mystery for over 70 years. It wasn’t until 1971 that British pharmacologist John Vane (who later received a Nobel Prize for his work) elucidated that aspirin functions by irreversibly acetylating and inhibiting cyclooxygenase (COX) enzymes, thereby blocking prostaglandin synthesis. This covalent modification prevents the enzyme from performing its catalytic function, leading to its pharmacological effects.
Similarly, penicillin, discovered by Alexander Fleming in 1928 and mass-produced during World War II, revolutionized the treatment of bacterial infections. Its unparalleled efficacy saved countless lives, making it one of the most impactful pharmaceutical discoveries. However, the intricate details of its covalent inhibition mechanism, specifically its interaction with bacterial transpeptidase enzymes (also known as penicillin-binding proteins, PBPs), were not fully understood until more than 50 years after its approval. Penicillin’s beta-lactam ring acts as a "suicide inhibitor," forming a stable covalent bond with a serine residue in the active site of bacterial transpeptidases, thereby preventing bacterial cell wall synthesis and leading to bacterial lysis.
These early successes, while transformative, laid bare a fundamental challenge: the drugs’ mechanisms were often poorly understood at the time of their clinical application. This lack of mechanistic insight, coupled with later observations of toxicity associated with certain reactive compounds, cast a long shadow over covalent drug development for decades.
The "No-Go" Era and the Rise of Caution
The initial enthusiasm for covalent drugs waned significantly as medicinal chemists became increasingly aware of the potential for indiscriminate reactivity and off-target toxicity associated with highly electrophilic compounds. Developers learned that while covalent bonds could confer potent and sustained pharmacological effects, poorly designed electrophiles could also bind non-selectively to a myriad of biological macromolecules—proteins, lipids, and even DNA—leading to cellular damage, genotoxicity, and, in some cases, severe adverse effects or cancer.
High-profile examples of drug-induced toxicities reinforced this perception. Acetaminophen, for instance, can form highly reactive electrophilic metabolites that, in overdose situations, deplete glutathione stores and bind covalently to hepatic proteins, leading to liver damage. Bromobenzene, a known hepatotoxin, and urushiol, the active allergen in poison ivy, are other classic examples of electrophiles causing adverse biological reactions through covalent adduct formation. These incidents solidified the notion that electrophilic compounds were "no-go" zones in drug development, pushing the industry towards the design of reversible, non-covalent inhibitors, which were generally perceived as safer due to their transient binding nature. This conservative shift effectively stalled covalent drug research for several decades, limiting the exploration of this potentially powerful therapeutic modality.
The Renaissance: Rational Design and "Soft Electrophiles"
The tide began to turn in the late 20th and early 21st centuries, driven by advancements in structural biology, computational chemistry, and a deeper understanding of target biology. Researchers recognized that the key to unlocking the therapeutic potential of covalent drugs lay in precision: designing "soft electrophiles" that could selectively target specific amino acid residues (most commonly cysteine) within the active sites of disease-relevant proteins, while minimizing off-target reactivity.
This paradigm shift moved away from indiscriminately reactive warheads towards rationally designed Targeted Covalent Inhibitors (TCIs). These new generation covalent drugs often utilize less reactive electrophilic groups, such as acrylamides, nitriles, and sulfonyl fluorides, strategically positioned to react with specific nucleophilic residues within the target protein. The success of this approach is epitomized by drugs like Ibrutinib, approved in 2013, which covalently binds to Cys481 in Bruton’s tyrosine kinase (BTK), revolutionizing the treatment of B-cell malignancies. Similarly, Osimertinib, an EGFR inhibitor approved for non-small cell lung cancer, forms a covalent bond with Cys797 in EGFR, effectively overcoming resistance mutations that plague earlier reversible inhibitors.
The distinction between a safe "soft electrophile" and a toxic one hinges on carefully tuning the electrophilicity of the warhead. A compelling example contrasting this fine line is the difference between sucralose and sulfur mustard. Both compounds contain alkyl chlorides. Sucralose, a widely used artificial sweetener, is safe for long-term consumption because its chloro-groups are sterically hindered and metabolically stable, making them non-reactive under physiological conditions. In stark contrast, sulfur mustard, a potent chemical weapon, readily alkylates biological molecules due to the high reactivity of its chloroethyl groups, causing severe cellular damage. This stark comparison underscores the critical importance of precise chemical design in the development of safe and effective covalent drugs.
This rational design approach has led to a boom in the field. To date, more than 110 covalent drugs have received regulatory approval across a diverse range of therapeutic areas, including infectious diseases (penicillin), gastrointestinal disorders (omeprazole), hepatitis C virus (telaprevir), and oncology (osimertinib, afatinib, ibrutinib). The market for covalent drugs is projected to grow significantly, reflecting their enhanced potency, prolonged duration of action, and ability to address previously "undruggable" targets. This growth is further supported by an increasing number of covalent drug candidates entering clinical trials, demonstrating the industry’s renewed confidence in this modality.
Navigating the Unique Pharmacokinetic Landscape
Despite the renewed success, the inherent nature of covalent binding introduces a unique set of pharmacokinetic challenges that differentiate these drugs from conventional reversible small-molecule inhibitors. Effectively characterizing and managing these properties is paramount for successful drug development and regulatory approval. Key considerations include protein binding beyond the target, complex clearance mechanisms, the potential for time-dependent inhibition (TDI) of CYP450 enzymes, and a distinct PK/PD relationship.
Protein Binding Beyond the Target Protein: A Double-Edged Sword
While selective covalent binding to the intended therapeutic target is the cornerstone of a covalent drug’s efficacy, these compounds can also interact with other proteins throughout the body. This off-target covalent binding can profoundly influence a drug’s distribution, metabolism, and potential for toxicity.
A common scenario involves covalent drugs forming stable adducts with abundant plasma proteins, such as human serum albumin (HSA). While this might appear similar to reversible protein binding, the irreversible nature of covalent binding means that the drug remains sequestered for the lifetime of the protein. This can significantly reduce the concentration of free, unbound drug in the plasma, potentially affecting its ability to reach the target site and exert its pharmacological effect. Conversely, in some cases, covalent binding to plasma proteins can serve as a "depot" for the drug, slowly releasing it over time. However, it can also lead to altered drug distribution, as protein-bound drugs may not readily cross biological membranes.
Beyond plasma, covalent drugs can also bind to tissue proteins, particularly in highly metabolically active organs like the liver. Such interactions can influence drug retention in tissues, alter local concentrations, and contribute to organ-specific toxicities if the off-target binding disrupts essential cellular functions. For instance, binding to hepatic enzymes or structural proteins could impair liver function, even if the primary target is elsewhere.
Advice for Developers: It is crucial for drug developers to meticulously characterize both on-target engagement and off-target reactivity. Advanced analytical techniques, such as mass spectrometry-based proteomics, can identify specific protein adducts in vitro and in vivo. Critically, high levels of off-target binding do not inherently equate to adverse effects; the biological consequence of such binding must be carefully assessed. The emphasis should be on optimizing the selectivity of the electrophilic warhead through rational design, ensuring it preferentially reacts with the intended target’s nucleophilic residue while minimizing promiscuous interactions with other proteins. This requires a deep understanding of protein structure, active site dynamics, and the precise chemical reactivity of the warhead.
Complex Clearance Mechanisms: Conjugation and Adduct Turnover
The clearance of covalent drugs presents another layer of complexity. Many covalent drugs, particularly those with electrophilic warheads, undergo metabolism through conjugation pathways, primarily via glutathione (GSH) and cysteine adduct formation. These pathways represent major routes of metabolic inactivation, converting the reactive electrophile into more hydrophilic and excretable species.
Glutathione S-transferases (GSTs) are a family of enzymes that catalyze the conjugation of electrophilic compounds to GSH, a ubiquitous tripeptide thiol. Genetic polymorphisms in GST enzymes can lead to significant inter-individual variability in drug metabolism and susceptibility to toxicity. Therefore, early assessment of non-enzymatic and GST-mediated reactivity is critical. This involves quantifying the rate of adduct formation with GSH and using recombinant GST isoforms, along with hepatocyte systems, to identify potential risks related to genetic variations in patient populations.
A prime example is Futibatinib, an irreversible FGFR1-4 inhibitor approved for FGFR2-rearranged cholangiocarcinoma. A mass-balance study involving 14C-futibatinib revealed that its primary metabolism involved O-demethylation and extensive glutathione conjugation. The major circulating metabolite was identified as a cysteinylglycine conjugate, representing approximately 13% of the circulating radioactivity. Further hepatocyte studies identified additional GSH, cysteine, glucuronide, and sulfate metabolites, highlighting the multifaceted nature of its clearance.
Beyond direct conjugation, covalent drugs can also be eliminated through the breakdown and clearance of drug-protein adducts, such as those formed with albumin or other tissue proteins. These protein-adduct mediated clearance mechanisms can account for a significant portion of total drug excretion and can contribute to a prolonged elimination phase, even if the free drug has a short plasma half-life. Understanding the kinetics of protein turnover and degradation is thus crucial for accurately predicting the overall clearance profile of a covalent drug.
Advice for Developers: Comprehensive in vitro and in vivo studies are necessary to fully characterize the metabolic fate of covalent drugs. This includes quantifying non-enzymatic and GST-mediated reactivity, ranking electrophile reactivity, and assessing polymorphism-related risks using various cellular and enzymatic systems. The identification and characterization of protein adducts using advanced mass spectrometry techniques are essential for understanding clearance pathways and predicting long-term exposure and potential toxicities.
Time-Dependent Inhibition (TDI) of CYP450 Enzymes: A Drug-Drug Interaction Risk
A significant concern for any new drug candidate is its potential to cause drug-drug interactions (DDIs), particularly through the modulation of cytochrome P450 (CYP450) enzymes, which are responsible for metabolizing a vast array of drugs. Studies have consistently shown that a substantial number of covalent drugs act as time-dependent inhibitors (TDIs) of at least one human CYP enzyme.
TDI occurs when a drug or its metabolite forms a stable, irreversible complex with the CYP enzyme, leading to a progressive loss of enzyme activity over time. This differs from reversible inhibition, where the enzyme activity recovers as the inhibitor dissociates. For covalent drugs, the electrophilic warhead or its metabolite can covalently bind to the heme iron or the apoprotein of the CYP enzyme, permanently deactivating it. This necessitates the synthesis of new enzyme molecules to restore metabolic capacity, leading to prolonged inhibition.
A study by Moghaddam MF et al. (2014) evaluated ten covalent drugs across multiple CYP isoforms and found that most exhibited time-dependent inhibition of at least one enzyme. This suggests that the potential for TDI is a prevalent characteristic among covalent inhibitors and poses a significant clinical risk for DDIs, as co-administered drugs metabolized by the inhibited CYP enzyme could reach toxic concentrations.
Advice for Developers: Early and thorough kinetic characterization of TDI (determining KI, kinact values) is paramount during preclinical development. Mechanistic modeling based on these kinetic parameters can help predict the clinical significance of potential DDIs and guide structural optimization to mitigate TDI risk. If TDI cannot be avoided, the data can inform dosing strategies, such as staggering administration times or adjusting dosages of co-administered drugs, to minimize adverse interactions. Regulatory agencies typically require robust TDI assessments for all new drug candidates.
The Decoupling of PK and PD: Rethinking Efficacy Metrics
Perhaps the most distinctive pharmacokinetic characteristic of covalent drugs is the decoupling of their plasma concentration (PK) from their pharmacological effect (PD). For traditional, reversible small-molecule drugs, continuous maintenance of drug concentrations above a certain therapeutic threshold in the blood is typically required to sustain the desired pharmacological effect. Plasma half-life and AUC (Area Under the Curve) are critical parameters for guiding dosing regimens.
However, covalent drugs operate differently. Once a covalent drug binds irreversibly to its target protein, it persistently occupies the active site. The duration of the pharmacological effect is then dictated not by the drug’s plasma half-life, but by the turnover rate of the target protein. The body must synthesize new, functional target protein molecules to restore the activity that was inhibited by the covalent drug. This means that even if the drug is rapidly cleared from the plasma, the therapeutic effect can persist for an extended period, often hours or even days, until sufficient new target protein has been produced.
A compelling illustration of this phenomenon comes from the irreversible BTK inhibitor CC-292 (now known as Tirabrutinib). Studies showed that plasma levels of CC-292 fell to near or below the lower limit of quantification within 24 hours post-dose. Yet, target occupancy of BTK remained high for up to 24 hours and only began to decline as new BTK protein was resynthesized. This clear decoupling highlights that optimizing plasma exposure, as defined by traditional PK metrics like AUC or Cmin, is less critical than achieving sufficient initial target engagement (Cmax-driven rapid engagement).
For Ibrutinib, as detailed in the illustrative image, its prolonged target occupancy is the structural basis for its sustained effect, despite its relatively short plasma half-life. The covalent bond formed between its acrylamide warhead and Cys481 in BTK ensures that the enzyme remains inhibited until new BTK is synthesized. This fundamental difference necessitates a re-evaluation of how efficacy is monitored and predicted for covalent drugs.
Advice for Developers: To address this unique PK/PD relationship, researchers must shift their focus from plasma drug concentrations to direct measures of target engagement and target turnover. This involves developing and utilizing robust pharmacodynamic biomarkers, such as target occupancy assays (e.g., using mass spectrometry or Western blotting to quantify drug-bound protein) or phenotypic assays that directly measure the biological consequence of target inhibition. Dosing strategies should prioritize achieving a rapid and high initial Cmax to saturate the target, rather than sustaining plasma levels. Modeling target occupancy directly, considering the target’s resynthesis rate, is crucial for guiding optimal dosing regimens and predicting clinical outcomes.
The Path Forward: Expertise and Precision in Covalent Drug Development
Covalent drugs represent a powerful and indispensable class of therapeutics, offering solutions to some of the most challenging medical needs, particularly in areas like oncology, infectious diseases, and autoimmune disorders. Their ability to confer enhanced potency, overcome resistance mechanisms, and provide prolonged pharmacological effects makes them highly attractive.
To fully capitalize on this potential, developers must embrace a comprehensive and nuanced understanding of their unique pharmacokinetic properties. This necessitates moving beyond conventional small-molecule drug development paradigms and adopting specialized strategies throughout the entire drug discovery and development lifecycle, from initial design to regulatory review. Covalent drug programs must demonstrate a commitment to deliberate, selective chemistry, underpinned by a deep mechanistic understanding of both efficacy and safety. This involves meticulous warhead design, rigorous characterization of reactivity and selectivity, and sophisticated modeling of PK/PD relationships that account for target turnover.
As technological advancements continue to accelerate—from high-throughput screening for covalent binders to advanced computational chemistry and AI-driven design platforms—the development of covalent drugs is poised for further expansion. Development teams equipped with the necessary expertise and experience to design, characterize, and model these unique mechanisms will be better positioned to efficiently and safely bring transformative therapies to patients, ultimately expanding the therapeutic arsenal against debilitating diseases. The future of medicine will undoubtedly feature an increasing number of intelligently designed covalent drugs, carefully crafted to deliver maximal benefit with minimal risk.
SOURCES:
- Moghaddam MF, Tang Y, O’Brien Z, Richardson SJ, Bacolod M, Chaturedi P, Apuy J, Kulkarni A. A proposed screening paradigm for discovery of covalent inhibitor drugs. Drug Metab Lett. 2014;8(1):19-30.
- Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, Witowski SR, Lounsbury H, Chaturvedi P, Mazdiyasni H, Zhu Z, Nacht M, Freed MI, Petter RC, Dubrovskiy A, Singh J, Westlin WF. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013 Aug;346(2):219-28.
AUTHOR BIO:
Author: Dr. Qigan Cheng, Sr. Study Director, DMPK Department, WuXi AppTec
Brief Bio: Dr. Qigan Cheng currently is a senior Study Director in DMPK Department of WuXi AppTec. He has over 10 years of research experience in the field of drug discovery and development and 6 years of research experience in the field of preclinical pharmacokinetic. He has successfully supported over 30 new drug IND applications globally.





Leave a Reply