
The landscape of modern medicine has been profoundly shaped by therapeutic agents that form stable, enduring bonds with their biological targets. From the venerable aspirin, approved in 1899, to the life-saving penicillin, a revolutionary breakthrough that emerged shortly thereafter, covalent drugs have consistently delivered transformative patient benefits. In recent years, a significant resurgence in the development and approval of new covalent drugs has rekindled hope for patients battling conditions previously considered intractable. However, this renaissance in covalent drug discovery is accompanied by a unique set of challenges, particularly concerning their distinct pharmacokinetic (PK) characteristics, which demand a specialized and meticulous approach during development.
A New Era for Targeted Therapies: The Resurgence of Covalent Drugs
The journey of covalent drugs has been marked by periods of both pioneering discovery and cautious skepticism. Early breakthroughs, while revolutionary, were largely serendipitous. The precise mechanisms by which drugs like aspirin exerted their effects were often not fully elucidated until decades after their widespread clinical use. For instance, aspirin’s covalent inhibition of cyclooxygenase (COX) activity, central to its anti-inflammatory and antiplatelet actions, remained poorly understood for nearly 70 years post-approval. Similarly, the covalent inhibition mechanism of penicillin, which disrupts bacterial cell wall synthesis by forming a stable bond with bacterial transpeptidase enzymes, took over half a century to be fully deciphered.
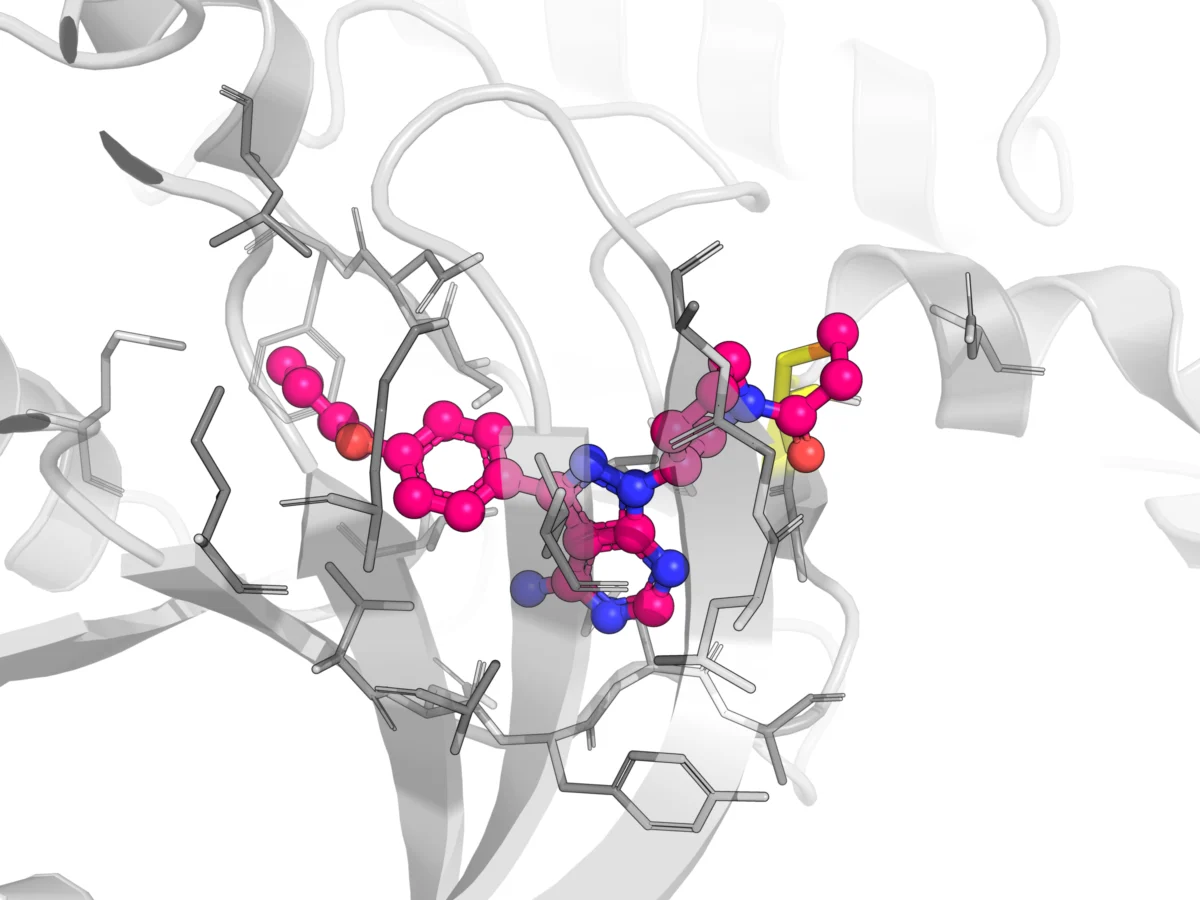
The foundational understanding that covalent drugs work by forming irreversible or quasi-irreversible bonds with specific residues on biological macromolecules, such as enzymes or receptors, thereby deactivating or altering their function, eventually emerged. This profound and often persistent engagement with the target is what grants many covalent drugs their exceptional efficacy and prolonged pharmacological effects.
Despite their early successes, the development of covalent drugs experienced a significant deceleration during the mid-20th century. This stagnation was primarily due to the prevailing perception that the electrophilic compounds inherent in their design were inherently unsafe. Early toxicological studies revealed that many reactive electrophiles could be metabolized into highly reactive intermediates that indiscriminately bound to a wide array of biomolecules—including proteins, lipids, and even DNA. Such promiscuous binding was linked to cellular damage, genotoxicity, and, in some cases, carcinogenicity. High-profile examples of severe toxicities associated with compounds like acetaminophen (in overdose), bromobenzene, and urushiol (the irritant in poison ivy) solidified the belief that electrophiles represented "no-go" zones for drug development. Consequently, medicinal chemistry largely shifted its focus towards the design of reversible, non-covalent inhibitors, effectively sidelining covalent mechanisms for decades.
The current landscape, however, reflects a dramatic paradigm shift. Modern covalent drug design now employs sophisticated "soft electrophiles," often utilizing chemical moieties such as acrylamides, nitriles, and sulfonyl fluorides as the reactive "warhead." These contemporary electrophiles are engineered to be far less reactive and significantly more selective than their predecessors, largely dispelling the earlier belief in their inherent promiscuity and unsafety. This evolution underscores the critical importance of carefully tuning the electrophilicity of the warhead during the drug discovery process to optimize target engagement while minimizing off-target reactivity and toxicity. A stark comparison illustrates this principle: sucralose, an artificial sweetener, and sulfur mustard, a chemical weapon, both contain alkyl chloride groups. Sucralose is safe for long-term consumption due to its precisely tuned reactivity, whereas mustard gas causes devastating alkylation injuries due to its extreme reactivity.
The success of these lower-reactivity warheads, particularly through the advent of rationally designed Targeted Covalent Inhibitors (TCIs), has ignited a boom in covalent drug development. To date, well over 110 covalent drugs have received regulatory approval across a diverse range of therapeutic areas. These include traditional examples like penicillin for infectious diseases and omeprazole for gastrointestinal disorders, alongside more recent innovations such as telaprevir for Hepatitis C Virus (HCV) and osimertinib, ibrutinib, and acalabrutinib in oncology. The field has matured to a point where researchers now employ sophisticated metrics, such as kinact/Ki (a measure of inactivation efficiency) and assessments of glutathione (GSH) conjugation and time-dependent inhibition (TDI) risk, to evaluate the reaction kinetics and potential off-target effects of these compounds. Yet, despite these advancements, the unique binding mechanisms of covalent drugs continue to present distinct and complex pharmacokinetic challenges that necessitate specialized attention throughout their development lifecycle.
Unpacking the Pharmacokinetic Paradox: Why Covalent Drugs Are Different
The fundamental principle of covalent binding imbues these drugs with pharmacokinetic properties that diverge significantly from those of conventional, reversible small-molecule therapeutics. Key considerations that demand particular scrutiny include their unique protein binding characteristics, specialized clearance mechanisms, propensity for CYP450 enzyme inhibition, and, most notably, a distinctive pharmacokinetic/pharmacodynamic (PK/PD) relationship.
Beyond the Binding Pocket: Navigating Off-Target Protein Interactions
While covalent binding to the intended therapeutic target protein is paramount for a drug’s efficacy, covalent compounds can also form adducts with other proteins throughout the body. These off-target interactions are not always benign and can significantly influence a drug’s distribution, metabolism, and potential for toxicity. For instance, many covalent drugs readily form stable adducts with human serum albumin (HSA), the most abundant protein in plasma. This extensive binding to HSA can effectively reduce the concentration of free, pharmacologically active drug in the plasma, thereby altering its volume of distribution and potentially impacting its clearance rate. While high levels of HSA binding might seem concerning, it does not inherently correlate with adverse effects. The challenge for drug developers lies in judiciously distinguishing between therapeutically relevant on-target engagement and undesirable off-target reactivity that could lead to toxicity. Furthermore, covalent drugs can bind to tissue proteins, particularly in highly metabolic organs like the liver. Such tissue retention can influence local drug concentrations, impact metabolic pathways, and contribute to organ-specific toxicities if not carefully managed. Therefore, rigorous optimization of the selectivity and finely tuned reactivity of these inhibitors is absolutely crucial to mitigate off-target effects and enhance the safety profile. Advanced analytical techniques, such as mass spectrometry-based proteomics, are increasingly employed to identify and quantify both on-target and off-target adducts in biological samples, providing critical insights into the drug’s selectivity profile.
The Metabolic Maze: Unique Clearance Pathways
Covalent drugs often undergo distinct clearance pathways compared to their non-covalent counterparts. A major route of metabolic inactivation for many electrophilic covalent drugs involves conjugation, particularly through the formation of glutathione (GSH) and cysteine adducts. Glutathione, a ubiquitous tripeptide, serves as a crucial cellular defense mechanism against electrophilic compounds, conjugating with them to form more water-soluble and excretable metabolites. Enzymes like glutathione S-transferases (GSTs) catalyze these reactions, but non-enzymatic conjugation can also occur. For example, Futibatinib, an irreversible FGFR1-4 inhibitor recently approved for FGFR2-rearranged cholangiocarcinoma, is primarily metabolized via O-demethylation and glutathione conjugation. A detailed mass-balance study of 14C-futibatinib revealed that a major circulating metabolite was a cysteinylglycine conjugate, accounting for approximately 13% of the circulating radioactive material. Further hepatocyte studies identified additional GSH, cysteine, glucuronide, and sulfate metabolites, underscoring the complexity of its metabolic fate.
Given these unique pathways, developers are advised to quantify both nonenzymatic and GST-mediated reactivity early in the discovery process. Ranking electrophile reactivity and utilizing recombinant GST isoforms and hepatocyte systems are essential steps to assess polymorphism-related risks, as genetic variations in GST enzymes can significantly impact individual drug metabolism and potential for toxicity. Beyond these direct conjugation routes, covalent drugs can also be eliminated through the breakdown and clearance of the drug-protein adducts themselves. For instance, drug-albumin adducts can be cleared through the normal turnover of albumin, potentially accounting for a substantial portion of the total drug excretion. This "adduct-mediated clearance" adds another layer of complexity to understanding the overall disposition of covalent drugs.
Drug-Drug Interaction Risks: Time-Dependent Inhibition of CYP450 Enzymes
A significant pharmacokinetic consideration for covalent drugs is their propensity for time-dependent inhibition (TDI) of cytochrome P450 (CYP450) enzymes, the primary enzymes responsible for drug metabolism in the liver. Numerous studies, including research by Moghaddam MF et al., have demonstrated that a majority of covalent drugs act as TDIs of at least one human CYP enzyme. This phenomenon, often termed mechanism-based inactivation (MBI), occurs when the drug or its metabolite covalently binds to the active site of the CYP enzyme, permanently deactivating it. Unlike reversible inhibition, TDI results in a progressive loss of enzyme activity over time and necessitates the synthesis of new enzyme protein for activity to recover.
The clinical implications of TDI are substantial, primarily leading to unpredictable and potentially dangerous drug-drug interactions (DDIs). If a covalent drug is a TDI of a CYP enzyme, it can impair the metabolism of co-administered drugs that are substrates for that same enzyme, leading to elevated plasma concentrations of the co-administered drug and an increased risk of toxicity. Conversely, if the co-administered drug is a prodrug requiring activation by the inhibited CYP enzyme, its therapeutic efficacy could be diminished. To mitigate these risks, early kinetic characterization (determining KI, the apparent inactivation constant, and kinact, the maximum inactivation rate) and mechanistic modeling are indispensable. These data help predict clinical DDI risk and can guide structural optimization efforts to reduce TDI potential or inform appropriate dosing strategies and drug interaction management plans. Regulatory agencies increasingly require comprehensive TDI assessment for all new drug candidates.
Decoupling Plasma Levels from Therapeutic Effect: The PK/PD Relationship
Perhaps the most distinctive pharmacokinetic characteristic of covalent drugs is the profound decoupling of their plasma concentration (PK) from their pharmacological effect (PD). For conventional, reversible small-molecule drugs, a sustained therapeutic effect typically requires maintaining drug concentrations within a specific therapeutic window in the blood. However, once a covalent drug forms its stable bond with the target protein, it persistently occupies the active site, exerting its effect long after the unbound drug has been cleared from the plasma. The recovery of target activity is then dictated not by the drug’s plasma half-life, but by the physiological turnover rate of the target protein—that is, the time required for the cell to synthesize new, functional target protein.
A compelling illustration of this phenomenon is observed with the irreversible Bruton’s tyrosine kinase (BTK) inhibitor, CC-292. In clinical studies, plasma levels of CC-292 rapidly fell to near or even below the lower limit of quantification within 24 hours post-dose. Despite this rapid plasma clearance, target occupancy of BTK remained remarkably high for up to 24 hours and only began to decline as new BTK protein was synthesized. This example unequivocally demonstrates that for covalent drugs, optimizing plasma exposure to maintain a steady-state concentration is far less critical than achieving sufficient initial target engagement to saturate the target protein. Therefore, developers are advised to shift their focus towards Cmax-driven rapid engagement and to directly model target occupancy, recognizing that pharmacodynamics are governed by target turnover kinetics rather than the conventional drug half-life. This fundamental difference often allows for less frequent dosing regimens compared to reversible inhibitors, contributing to improved patient adherence and convenience.
Advancing Development: Strategies for Success
To fully harness the transformative potential of covalent drugs, developers must adopt a sophisticated and multidisciplinary approach that integrates a deep understanding of their unique pharmacokinetic properties throughout the entire drug development and regulatory review process. This necessitates:
- Rational Design and Mechanistic Understanding: Moving beyond serendipity, modern covalent drug development emphasizes rational design principles. This includes precise tuning of the electrophilic warhead’s reactivity and selectivity to ensure potent on-target binding while minimizing off-target interactions. A thorough mechanistic understanding of safety, exposure, and efficacy is paramount.
- Advanced Analytical Techniques: Characterizing the complex behavior of covalent drugs requires state-of-the-art analytical methods. Techniques such as mass spectrometry are essential for identifying and quantifying drug-protein adducts, both on-target and off-target, in various biological matrices. Imaging techniques like PET scans with radiolabeled covalent inhibitors can provide in-vivo quantification of target occupancy in specific tissues, offering invaluable insights into drug distribution and engagement.
- Specialized Expertise and Collaboration: The intricacies of covalent pharmacology demand specialized expertise in medicinal chemistry, pharmacology, toxicology, and DMPK (Drug Metabolism and Pharmacokinetics). Collaborations between academic institutions, pharmaceutical companies, and Contract Research Organizations (CROs) like WuXi AppTec, which possess extensive experience in preclinical pharmacokinetic studies and IND applications, are becoming increasingly vital. These partnerships facilitate the efficient design, characterization, and modeling of these unique mechanisms, accelerating the delivery of transformative therapies to patients safely.
- Tailored Regulatory Frameworks: Regulatory bodies worldwide are adapting their guidelines to accommodate the unique properties of covalent drugs. Developers must be prepared to provide comprehensive data on target engagement, TDI potential, and adduct formation, ensuring that their programs demonstrate deliberate, selective chemistry with a clear mechanistic understanding of both safety and exposure.
The Horizon of Covalent Therapeutics: Broader Impact and Future Directions
Covalent drugs are addressing some of the most critical unmet needs in modern therapeutics, particularly in oncology, where they offer sustained inhibition of disease-driving mutations, often overcoming resistance mechanisms. Beyond cancer, their application is expanding into autoimmune diseases, infectious diseases, and potentially even neurodegenerative disorders, where persistent target modulation can be highly advantageous.
The principles underlying covalent drug design are also paving the way for next-generation therapeutic modalities. For instance, Proteolysis-Targeting Chimeras (PROTACs) and other targeted protein degraders often leverage covalent or quasi-covalent interactions to bring an E3 ligase into close proximity with a target protein, leading to its ubiquitination and subsequent degradation. This innovative approach offers the potential to "drug the undruggable" by targeting proteins previously considered inaccessible to traditional inhibitors. The ongoing advancements in chemical biology, computational chemistry, and structural biology will continue to accelerate covalent drug development. Development teams equipped with the necessary expertise and experience to navigate the complexities of covalent mechanisms will be exceptionally well-positioned to bring these highly effective and often transformative therapies to patients with greater efficiency and enhanced safety. The future of medicine will undoubtedly feature an increasing number of intelligently designed covalent drugs, offering durable solutions to persistent health challenges.







Leave a Reply