Researchers at the University of California, Riverside (UCR) have unveiled a provocative new theory regarding the molecular origins of Alzheimer’s disease, suggesting that the condition may be triggered by a direct competition between two key proteins inside nerve cells. The study, published in the Proceedings of the National Academy of Sciences (PNAS) Nexus, challenges the long-standing "amyloid cascade hypothesis," which posits that the accumulation of extracellular amyloid-beta (a-beta) plaques is the primary driver of the disease. Instead, the UCR team proposes that Alzheimer’s begins when a-beta enters the interior of the neuron and physically displaces the tau protein from microtubules, the structural "highways" essential for cellular survival.
This shift in perspective arrives at a critical juncture in neurology. For decades, the pharmaceutical industry has invested billions of dollars into therapies designed to clear a-beta plaques from the brain, yet these treatments have yielded remarkably modest clinical benefits. By shifting the focus from the debris outside the cell to the structural integrity inside the cell, the UCR researchers may have identified the "missing link" that explains why clearing plaques often fails to stop cognitive decline.
The Evolution of Alzheimer’s Research: From Plaques to Processes
The history of Alzheimer’s research has been defined by a search for a single, unifying cause. Since Alois Alzheimer first described the "plaques and tangles" in the brain of a deceased patient in 1906, scientists have debated which of these two hallmarks is the true culprit.
The amyloid-beta protein, which forms the plaques, became the primary focus in the late 20th century. This was reinforced by the discovery of rare genetic mutations that lead to early-onset Alzheimer’s by increasing a-beta production. However, the "tau hypothesis" also gained ground, noting that the accumulation of tau tangles—twisted fibers of protein inside the neurons—correlates much more closely with the actual severity of dementia and cognitive impairment than plaque density does.
Despite these observations, the two proteins were often studied in isolation. "In addition to having dementia, Alzheimer’s diagnosis requires both a-beta and tau buildup in the brain," explained Ryan Julian, a professor of chemistry at UCR and the lead author of the study. "But many labs focus on the role of one and ignore the other." The UCR study seeks to bridge this gap by demonstrating that a-beta and tau are not merely co-conspirators in the brain’s decline, but are actually direct competitors for the same biological real estate.
The Intracellular Battle for Microtubules
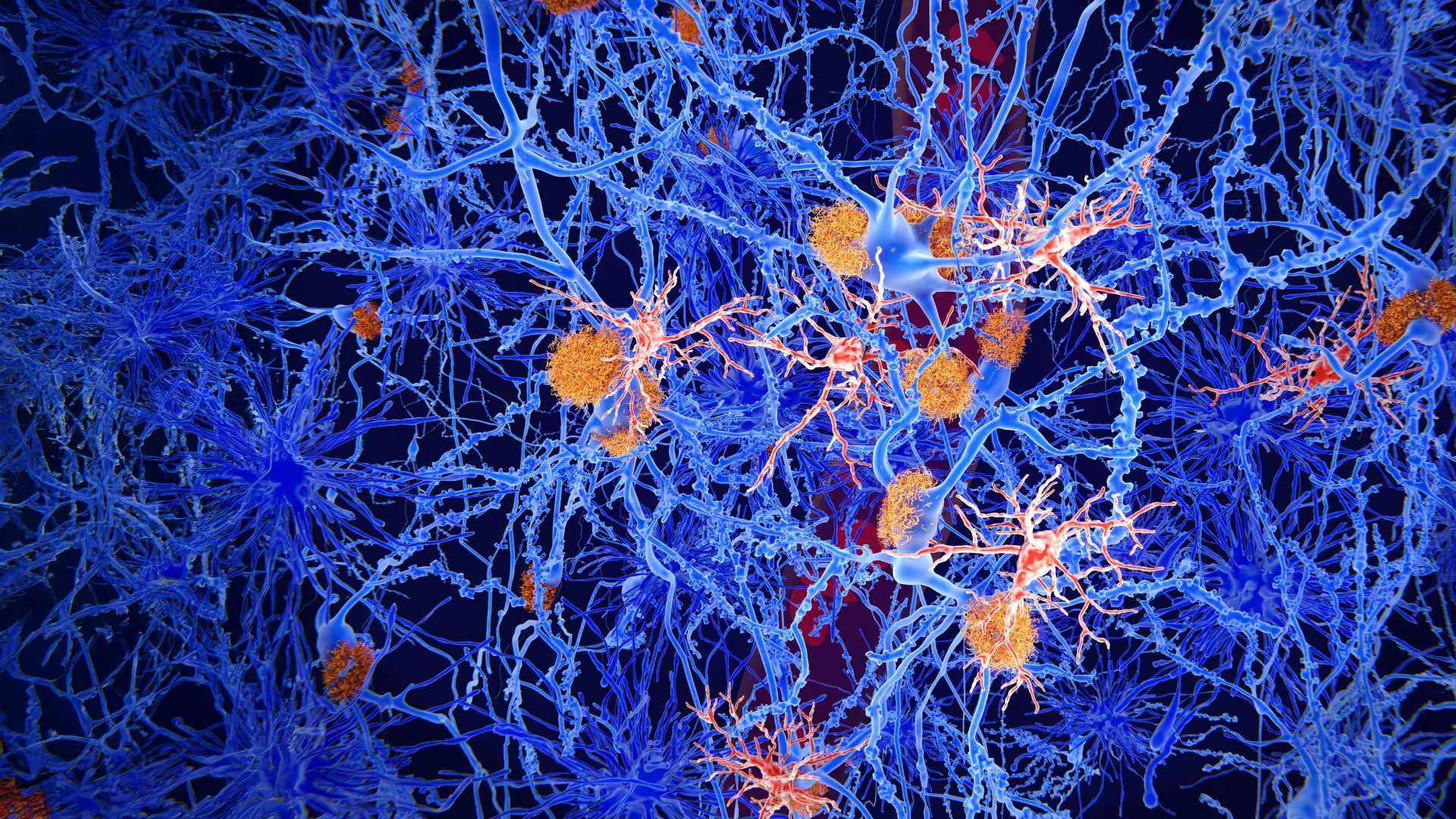
At the heart of the UCR study is the microtubule, a microscopic tube-like structure that serves two vital functions: providing structural scaffolding for the neuron and acting as a track for the transport of nutrients, neurotransmitters, and waste. For these tracks to remain stable and functional, they require the tau protein, which binds to the microtubules and keeps them from falling apart.
The research team noticed a striking similarity between a specific section of the tau protein and the a-beta protein. Both proteins share a similar size and structural configuration in the regions where they interface with other molecules. This observation led the team to hypothesize that a-beta might be capable of binding to the same spots on the microtubules that tau occupies.
To test this, the researchers utilized advanced chemical techniques, including the attachment of fluorescent markers to a-beta proteins. By monitoring the movement and light emission of these proteins under a microscope, they were able to observe the binding process in real-time. The results were definitive: a-beta binds to microtubules with an affinity similar to that of tau.
"Our work shows amyloid beta and tau compete for the same binding sites on microtubules, and that a-beta can prevent tau from functioning correctly," Julian stated. When internal a-beta levels rise, the protein effectively "kicks" the tau protein off the microtubule. This displacement has a twofold catastrophic effect: the microtubule loses its stability and begins to break down, and the displaced tau protein, now floating freely in the cytoplasm, begins to clump together into the toxic tangles characteristic of the disease.
A Chronology of Cellular Failure
The UCR model provides a logical timeline for the progression of Alzheimer’s that reconciles several previously confusing observations.
- Initial Accumulation: In a healthy, young brain, a-beta is produced and cleared efficiently. However, as a person ages, or due to genetic factors, the clearance mechanisms begin to slow down.
- Intracellular Infiltration: While much of the a-beta forms plaques outside the cells, a significant portion remains inside the neuron.
- Microtubule Displacement: As internal a-beta concentrations reach a threshold, they begin to outcompete tau for microtubule binding sites.
- Transport Collapse: The neuron’s internal transport system fails. Essential materials can no longer reach the synapses, leading to a loss of communication between neurons.
- Tau Tangle Formation: The displaced tau protein migrates to other parts of the cell, such as the cell body (soma), where it aggregates into neurofibrillary tangles.
- Cell Death: Deprived of nutrients and structural integrity, the neuron eventually dies, leading to the brain atrophy seen in advanced Alzheimer’s.
This "inside-out" theory explains why some patients can have significant plaque buildup in their brains but remain cognitively normal. If the a-beta remains outside the cells and does not interfere with the internal microtubules, the neurons can continue to function. It is only when the internal competition begins that the clinical symptoms of dementia manifest.
The Role of Autophagy and Aging
A critical component of the UCR study is the emphasis on autophagy—the cell’s natural "recycling" process. Autophagy is responsible for identifying, breaking down, and removing damaged proteins, including a-beta.
Data from various longitudinal studies suggest that autophagy efficiency declines significantly with age. When this "garbage disposal" system fails, proteins that should be removed instead linger inside the cell. The UCR researchers argue that the age-related decline in autophagy is likely the catalyst that allows internal a-beta levels to rise high enough to displace tau.
This connection to autophagy also aligns with recent epidemiological data. Some studies have indicated that long-term use of certain medications, such as lithium, may be associated with a lower risk of developing Alzheimer’s. Interestingly, lithium has been shown in separate laboratory studies to both stimulate autophagy and help stabilize microtubules. By strengthening the microtubule and helping the cell clear out competing a-beta, lithium—or drugs with similar mechanisms—could potentially interrupt the disease process at its earliest stages.
Supporting Data and the Failure of Traditional Therapies
The UCR findings provide a scientific framework for understanding the high failure rate of Alzheimer’s drugs over the last two decades. Between 2003 and 2021, no new drugs for Alzheimer’s were approved by the FDA, despite hundreds of clinical trials. More recent approvals, such as aducanumab and lecanemab, target the removal of external plaques. While these drugs have shown an ability to clear amyloid from the brain, their impact on slowing cognitive decline has been described by many in the medical community as "modest" or "incremental."
According to the UCR theory, these drugs may be "cleaning the yard while the house is on fire." If the primary damage is occurring inside the cell due to microtubule displacement, removing the external plaques may be insufficient to restore the neuron’s internal transport system once it has already collapsed.
Furthermore, statistics from the Alzheimer’s Association highlight the urgency of finding a more effective intervention. Currently, more than 6 million Americans are living with Alzheimer’s, a number projected to rise to nearly 13 million by 2050. The global economic burden is estimated at over $1 trillion annually. The UCR research suggests that the most effective future treatments may not be those that simply remove protein clumps, but those that protect the microtubule-tau bond or enhance the cell’s internal cleaning mechanisms.
Implications for Future Treatment and Diagnosis
The implications of this study for future drug development are profound. If the competition for microtubule binding is indeed the "trigger" for the disease, several new therapeutic avenues open up:
- Microtubule Stabilizers: Drugs that reinforce the bond between tau and microtubules, or that make the microtubule resistant to displacement by a-beta, could preserve neuronal function even in the presence of amyloid.
- Autophagy Enhancers: Developing compounds that can "re-boot" the cell’s recycling system in older adults could prevent the accumulation of internal a-beta before it reaches dangerous levels.
- Intracellular Targeting: Future biologics may need to be designed to enter the neuron more effectively to neutralize a-beta at the site of the microtubule, rather than just clearing it from the surrounding brain tissue.
Professor Ryan Julian believes this new model brings a much-needed sense of cohesion to the field. "This idea helps make sense of many results that previously seemed unrelated," he noted. "It gives us a clearer picture of what may be going wrong inside neurons and where new treatments might start."
Conclusion and Scientific Outlook
The UCR study represents a significant pivot in the conceptualization of Alzheimer’s disease. By identifying a physical interaction between a-beta and tau at the level of the microtubule, researchers have moved beyond observing the "end-state" debris of the disease and into the mechanics of cellular failure.
While further research is required to confirm these interactions in human clinical settings, the study provides a compelling explanation for the limitations of current therapies and a roadmap for the next generation of Alzheimer’s research. As the global population ages, the transition from plaque-centric models to structural and functional cellular models may prove to be the breakthrough necessary to finally stall the progression of this devastating neurodegenerative condition.